Two simple precursor 2,2′-bisoxazoline complexes of ruthenium
Pankaj K.
Pal
a,
Michael G. B.
Drew
b and
Dipankar
Datta
*a
aDepartment of Inorganic Chemistry, Indian Association for the Cultivation of Science, Calcutta 700 032. India. E-mail: icdd@mahendra.iacs.res.in
bDepartment of Chemistry, University of Reading, Whiteknights, Reading, UK RG6 6AD
First published on 7th January 2002
Abstract
Syntheses, X-ray crystal structures and some electrochemical properties of RuIIL2Cl2, RuIIIL(dimethylformamide)Cl3 and [RuIIL2Cl2]PF6, where L is 4,4,4′,4′-tetramethyl-2,2′-bisoxazoline, are described. It is found that the first two complexes serve as very good precursors to synthesise a host of mixed-ligand complexes of Ru(II) and Ru(III) containing L.
In the last decade, various substituted chiral bisoxazolines of the types 1 and 2 have emerged as an efficient class of ligands in metal-catalysed asymmetric syntheses.1–4 While a number of metal complexes of 2 (Evans's ligand) have been prepared and characterised structurally in various contexts,1–5 very little attention has been paid to the transition metal chemistry of 1. This is possibly because in most cases, the bisoxazolines of type 2 have been found to be superior to those of type 1 in producing a greater enantiomeric excess. In an extensive review on the metal complexes of oxazolines5 that appeared in 1999, Gómez et al. write that “unfortunately, to date, no X-ray structures containing chelate” 1 “have been determined.” After the appearance of this review, Walther et al. have reported the X-ray crystal structures of octahedral Ni(1a)(acetylacetonate)2 and square planar Ni(1a)(mes)(Br) (mes≡mesityl) and Pd(1a)(mes)2 in connection with their studies on the catalytic transformations brought about by Pd(II) and Ni(II) complexes.6 Recently, we have undertaken a project to develop the transition metal chemistry of the bisoxazolines of type 1. Elsewhere we have reported7 the photophysics and structure of a copper(I) dimer of a derivative of 1. Herein we make a preliminary report of our studies on its ruthenium chemistry. The syntheses, structures and some properties of two simple ruthenium complexes of 1a, from which a host of mixed-ligand complexes of ruthenium(II) and ruthenium(III) can be prepared, are described here.
Reaction of 1 mol of RuCl3·3H2O with 2 mol of the ligand 1a in hot dimethylformamide (DMF) in the presence of 0.5 mol of metallic zinc and an excess of the chloride ion leads to the isolation of Ru(1a)2Cl2 in 25% yield; after allowing the filtrate to stand in air for several days, Ru(1a)(DMF)Cl3 is obtained in 40% yield. The use of zinc metal in our synthesis is novel. For example, Ru(bpy)2Cl2 or Ru(phen)2Cl2 (bpy≡2,2′-bipyridine; phen≡1,10-phenanthroline) are synthesised in a similar manner without adding metallic zinc to the reaction mixture.8,9 The role of zinc here is to reduce ruthenium(III) to ruthenium(II); no trace of metallic zinc can be found in the crude product of Ru(1a)2Cl2. Incidentally, no bpy or phen analogue of Ru(1a)(DMF)Cl3 is known.
The structures of Ru(1a)2Cl2 and Ru(1a)(DMF)Cl3 as determined by X-ray crystallography are shown in Fig. 1 and 2, respectively. The ligand 1a, which is a non-aromatic 1,4-diimine, is found to bind ruthenium through the N ends and the DMF molecule in Ru(1a)(DMF)Cl3 binds through the O atom. In both the complexes, the metal atom has a slightly distorted octahedral environment; the distortion is mainly caused by the chelate bites [77.6(2)° in Ru(1a)2Cl2 and 78.0(4)° in Ru(1a)(DMF)Cl3]. The chlorine atoms in Ru(1a)2Cl2 are mutually cis and those in Ru(1a)(DMF)Cl3 span a meridian. The Ru–Cl and the Ru–N bond lengths in Ru(1a)(DMF)Cl3 are somewhat shorter than those in Ru(1a)2Cl2, indicative of the differences between the radii of Ru(III) and Ru(II). The bidentate ligands are slightly distorted from planarity with deviations of individual atoms from the 5-membered ring planes being less than 0.15 Å; the two rings intersect at angles of 5.2 and 8.1° in Ru(1a)2Cl2 and 1.9° in Ru(1a)(DMF)Cl3.
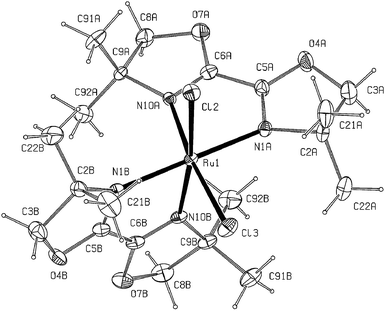 | ||
| Fig. 1 X-Ray crystal structure of Ru(1a)2Cl2 with ellipsoids at 15% probability. Selected bond distances (Å) and angles (°): Ru1–N1A 2.098(5), Ru1–N1B 2.102(5), Ru1–N10A 2.091(5), Ru1–N10B 2.082(4), Ru1–Cl2 2.421(3), Ru1–Cl3 2.416(3), N1A–Ru1–N1B 178.2(2), N10A–Ru1–Cl3 172.2(1), N10B–Ru1–Cl3 88.0(2), N1A–Ru1–Cl3 94.8(2), N1B–Ru1–Cl3 85.8(1). | ||
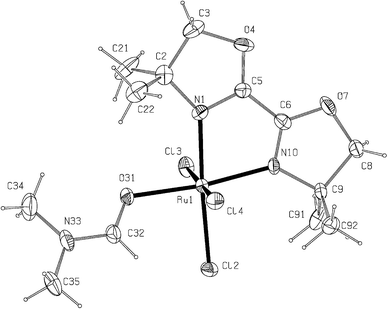 | ||
| Fig. 2 X-Ray crystal structure of Ru(1a)(DMF)Cl3 with ellipsoids at 15% probability. Selected bond distances (Å) and angles (°): Ru1–N1 2.063(10), Ru1–N10 2.061(9), Ru1–Cl2 2.367(4), Ru1–Cl3 2.330(4), Ru1–Cl4 2.353(4), Ru1–O31 2.109(8), N1–Ru1–Cl2 176.6(3), N10–Ru1–Cl2 98.6(3), Cl2–Ru1–Cl4 91.4(2), Cl2–Ru1–Cl3 91.3(2), Cl3–Ru1–Cl4 176.7(1), N1–Ru1–O31 94.9(4), N10–Ru1–O31 172.9(4), O31–Ru1–Cl2 88.5(3). | ||
In the 1H NMR spectrum of 1a in CDCl3, the methyl protons appear as a singlet at 1.37 ppm and the methylene ones as another singlet at 4.13 ppm. The presence of a twofold axis of symmetry in 1a is reflected in its 13C NMR spectrum also, in which four types of C atoms are indicated [methyl C, 28.42; C(CH3)2, 68.80; methylene C, 80.10; imino C, 153.67 ppm with reference to TMS]. However, this symmetry is lost in Ru(1a)2Cl2, as revealed by its 13C NMR spectrum in CDCl3 where 10 distinct resonances are observed (see Experimental), indicating that none of the ten carbon atoms in a particular ligand fragment in the complex are magnetically equivalent. The 1H NMR spectrum of Ru(1a)2Cl2 is very rich and we have not yet been able to decipher it.
The electrochemical behaviour of Ru(1a)2Cl2 and Ru(1a)(DMF)Cl3 has been examined by cyclic voltammetry at a glassy carbon electrode in acetonitrile under N2 atmosphere. Ru(1a)2Cl2 shows (Fig. 3) a quasireversible RuIII/II couple at 0.18 V vs. SCE (saturated calomel electrode). This RuIII/II couple appears at −0.34 V vs. SCE in Ru(1a)(DMF)Cl3 (Fig. 3). On the positive side of SCE, Ru(1a)(DMF)Cl3 displays a quasireversible RuIV/III couple at 1.22 V vs. SCE. For comparison, we note that the RuIII/II couple in cis-Ru(bpy)2Cl2 and cis-Ru(phen)2Cl2 appears10,11 respectively at 0.31 and 0.33 V vs. SCE in acetonitrile at a platinum electrode.
Because of the very low potential of the RuIII/II couple, Ru(1a)2Cl2 is easily oxidised by air. We have found that it is possible to isolate [Ru(1a)2Cl2]PF6, the ruthenium(III) counterpart of Ru(1a)2Cl2, just by refluxing a methanolic solution of Ru(1a)2Cl2 in air and by subsequent addition of excess NH4PF6. It should be noted that an oxidising agent like chlorine is needed in the synthesis of [Ru(bpy)2Cl2]PF6 from Ru(bpy)2Cl2.12 In X-ray crystallography, [Ru(1a)2Cl2]PF6 is found to consist of discrete [Ru(1a)2Cl2]+ cations and PF6− anions with both the cation (Fig. 4) and anion having crystallographic Cs symmetry. The chlorine atoms in [Ru(1a)2Cl2]+ are found to maintain the cis configuration. While the Ru–Cl bonds in [Ru(1a)2Cl2]PF6 are significantly shorter than those in Ru(1a)2Cl2, the Ru–N bonds in the complexes Ru(1a)2Cl2 and [Ru(1a)2Cl2]PF6 are comparable. The two 5-membered rings of 1a in [Ru(1a)2Cl2]PF6 intersect at an angle of 5.6°. In cyclic voltammetry at a glassy carbon electrode in acetonitrile under N2 atmosphere, the RuIII/II couple in [Ru(1a)2Cl2]PF6 is found to be lower by 40 mV in potential and much less reversible than that in Ru(1a)2Cl2 (Fig. 3). In this context, we mention that the RuIII/II couple in [Ru(bpy)2Cl2]+ is reported13 to be lower by 20 mV in potential but somewhat more reversible than that in Ru(bpy)2Cl2.
![The cyclic voltammograms of Ru(1a)2Cl2
(○, c = 1.09 mM), Ru(1a)(DMF)Cl3
(----, c = 1.38 mM) and [Ru(1a)2Cl2]PF6
(––, c = 0.42 mM) in acetonitrile (0.1 M in tetraethylammonium perchlorate) at a glassy carbon electrode. Scan rate ν = 50 mV s−1. Under the same experimental conditions, the ferrocene–ferrocenium couple appears at 0.35 V vs. SCE with a peak-to-peak separation of 80 mV at ν = 50 mV s−1.](/image/article/2002/NJ/b109294b/b109294b-f3.gif) | ||
Fig. 3 The cyclic voltammograms of Ru(1a)2Cl2
(○, c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =1.09 mM), Ru(1a)(DMF)Cl3
(----, c=1.38 mM) and [Ru(1a)2Cl2]PF6
(––, c=0.42 mM) in acetonitrile (0.1 M in tetraethylammonium perchlorate) at a glassy carbon electrode. Scan rate ν=50 mV s−1. Under the same experimental conditions, the ferrocene–ferrocenium couple appears at 0.35 V vs. SCE with a peak-to-peak separation of 80 mV at ν=50 mV s−1. =1.09 mM), Ru(1a)(DMF)Cl3
(----, c=1.38 mM) and [Ru(1a)2Cl2]PF6
(––, c=0.42 mM) in acetonitrile (0.1 M in tetraethylammonium perchlorate) at a glassy carbon electrode. Scan rate ν=50 mV s−1. Under the same experimental conditions, the ferrocene–ferrocenium couple appears at 0.35 V vs. SCE with a peak-to-peak separation of 80 mV at ν=50 mV s−1. | ||
![X-Ray crystal structure of [Ru(1a)2Cl2]PF6 with ellipsoids at 15% probability. Selected bond distances (Å) and angles (°): Ru1–N1 2.113(4), Ru1–N10 2.101(3), Ru1–Cl1 2.329(3), N1–Ru1–N10 77.2(2), N1–Ru1–N1 178.7(2), N10–Ru1–Cl1 89.1(1), 168.5(1).](/image/article/2002/NJ/b109294b/b109294b-f4.gif) | ||
| Fig. 4 X-Ray crystal structure of [Ru(1a)2Cl2]PF6 with ellipsoids at 15% probability. Selected bond distances (Å) and angles (°): Ru1–N1 2.113(4), Ru1–N10 2.101(3), Ru1–Cl1 2.329(3), N1–Ru1–N10 77.2(2), N1–Ru1–N1 178.7(2), N10–Ru1–Cl1 89.1(1), 168.5(1). | ||
In Scheme 1 we show how14 a number of mixed-ligand complexes of ruthenium(II) and ruthenium(III) containing 2,2′-bisoxazoline can be synthesised by replacing the chlorine atoms in Ru(1a)2Cl2 and the DMF molecule in Ru(1a)(dmf)Cl3. Thus, we envisage a ruthenium chemistry of 2,2′-bisoxazoline that will be as rich as that of bipyridine.
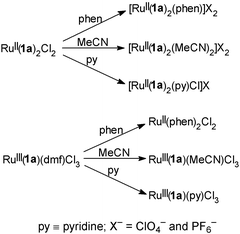 | ||
| Scheme 1 | ||
Experimental
Methods and materials
The ligand 1a (4,4,4′,4′-tetramethyl-2,2′-bisoxazoline) was synthesised by following a reported procedure.15 Microanalyses were performed on a Perkin–Elmer 2400II elemental analyser. Molar conductance was determined by a Systronics (India) direct reading conductivity meter (model 304). FTIR spectra (KBr disc) were recorded on a Nicolet Magna-IR spectrophotometer (Series II), UV-VIS spectra on a Shimadzu UV-160A spectrophotometer and NMR spectra (in CDCl3) by a Brucker DPX300 spectrometer. Cyclic voltammetry was performed using an EG&G PARC electrochemical analysis system (model 250/5/0) in purified acetonitrile under a dry nitrogen atmosphere in conventional three-electrode configurations. A planar EG&G PARC G0229 glassy carbon milli electrode was used as the working electrode. Magnetic susceptibility was determined at room temperature by a PAR 155 vibrating sample magnetometer. The magnetometer was calibrated with Hg[Co(SCN)4] and the susceptibility data were corrected for diamagnetism using Pascal's constants. In the calculation of the magnetic moments of the two ruthenium(III) complexes, Ru(1a)(DMF)Cl3 and [Ru(1a)2Cl2]PF6, we have ignored the temperature-independent paramagnetism (TIP) of the Ru3+ ion.Syntheses
°C until a microcrystalline compound started appearing (almost 1 h was needed). The solution was then cooled to room temperature, 10 ml of acetone was added and the solution kept in the refrigerator overnight. The next day the violet crystalline compound that had appeared was filtered off, washed with 10 ml of water and dried in vacuo over fused CaCl2. It was recrystallised from a dichloromethane–hexane mixture. Yield 0.15 g (25%). Anal. calcd for C20H32Cl2N4O4Ru: C, 42.53; H, 5.72; N, 9.92; found: C, 42.65; H, 5.66; N, 10.01%. UV/VIS (CH3CN)
λmax/nm (ε/dm3
mol−1 cm−1): 532 (8000), 464 (4300), 247 (6500), 220 (10,100). 13C NMR (300 MHz, CDCl3, TMS): δ 15.23, 25.07, 27.22, 29.57 (four methyl C), 65.81, 71.44 (two alkyl quaternary C), 82.52, 83.88 (two methylene C), 155.10, 157.59 (two imino C).
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O). μ/μB: 2.07 (at 300 K). UV/VIS (CH3CN)
λmax/nm (ε/dm3 mol−1 cm−1): 235 (10,500), 324 (2250), 379 (5900).
O). μ/μB: 2.07 (at 300 K). UV/VIS (CH3CN)
λmax/nm (ε/dm3 mol−1 cm−1): 235 (10,500), 324 (2250), 379 (5900).
X-Ray crystallography
Single crystals of Ru(1a)2Cl2, Ru(1a)(DMF)Cl3 and [Ru(1a)2Cl2]PF6 were grown by direct diffusion of hexane into a dilute dichloromethane solution of the respective complexes. Data were collected with Mo-Kα radiation using the MARresearch Image Plate System. The crystals were positioned at 70 mm from the Image Plate. One hundred frames were measured at 2° intervals with a counting time of 2 min. Data analyses were carried out with the XDS program.16 The structures were solved using direct methods with the SHELXS-86 program.17 In all the structures the non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms bonded to carbon were included in geometric positions and given thermal parameters equivalent to 1.2 times those of the atom to which they were attached. The structures were refined on F2 using SHELXL-93.18=564.47, monoclinic, space group P21/a, a=15.42(2), b=9.832(14), c=16.076(19)
Å, β=96.67(1)°, U=2421(6)
Å3, Z=4, Dc=1.549 g cm−3, μ=0.901 mm−1, 6185 reflections collected, 3926 unique, R(int)=0.031, final residuals: R1=0.0457 and wR2=0.1207 for observed data, R1=0.0857
and wR2=0.1429 for all data. Ru(1a)(DMF)Cl3: C13H23Cl3N3O3Ru, Mw=476.76, orthorhombic, space group P212121, a=8.607(14), b=14.48(2), c=15.73(2)
Å, U=1960(5)
Å3, Z=4, Dc=1.615 g cm−3, μ=1.223 mm−1, 2046 unique reflections collected, final residuals: R1=0.0581 and wR2=0.1598 for observed data, R1=0.0994 and wR2=0.1835 for all data. [Ru(1a)2Cl2]PF6:
C20H32Cl2N4O4RuPF6, Mw=709.44, orthorhombic, space group P22121, a=8.545(12), b=12.001(16), c=14.002(20)
Å, U=1436(4)
Å3, Z=4, Dc=1.641 g cm−3, μ=0.860 mm−1, 5008 reflections collected, 2750 unique, R(int)=0.027, final residuals: R1=0.0354 and wR2=0.0780 for observed data, R1=0.0479 and wR2=0.0832 for all data.
CCDC reference numbers 174511–174513. See http://www.rsc.org/suppdata/nj/b1/b109294b/ for crystallographic data in CIF or other electronic format.
Acknowledgements
M. G. B. D. thanks EPSRC and University of Reading for funds for the Image Plate System. D. D. thanks the Department of Science and Technology, New Delhi, India for financial support.References
- F. Fache, E. Schultz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS.
- D. A. Evans, K. A. Scheidt, N. Johnston and M. C. Willis, J. Am. Chem. Soc., 2001, 123, 4480 CrossRef CAS.
- R. Annuziata, M. Beneglia, M. Cinquini, F. Cozzi and M. Pitillo, J. Org. Chem., 2001, 66, 3160 CrossRef CAS.
- M. Gómez, G. Muller and M. Rocamora, Coord. Chem. Rev., 1999, 193–195, 769 CrossRef CAS.
- D. Walther, T. Döhler, K. Heubach, O. Klobes, B. Schweder and H. Görls, Z. Anorg. Allg. Chem., 1999, 625, 923 CrossRef CAS.
- G. K. Patra, I. Goldberg, S. K. Chowdhury, B. C. Maiti, A. Sarkar, P. R. Bangal, S. Chakravorti, N. Chattopadhyay, D. A. Tocher, M. G. B. Drew, G. Mostafa, S. Chowdhury and D. Datta, New J. Chem., 2001, 25, 1371 RSC.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334 CrossRef CAS.
- P. Bonneson, J. L. Walsh, W. T. Pennington, A. W. Cordes and B. Durham, Inorg. Chem., 1983, 22, 1761 CrossRef CAS.
- B. P. Sullivan, D. J. Salmon, T. J. Meyer and J. Peedin, Inorg. Chem., 1979, 18, 3369 CrossRef CAS.
- T. Matsumura-Inoue, I. Ikemoto and Y. Umezawa, J. Electroanal. Chem., 1986, 209, 135 CrossRef CAS.
- J. N. Braddock and T. J. Meyer, Inorg. Chem., 1973, 12, 723 CrossRef CAS.
- T. R. Weaver, T. J. Meyer, S. A. Adeyemi, G. M. Brown, R. P. Eckberg, W. E. Hatfield, E. C. Johnson, R. W. Murray and D. Untereker, J. Am. Chem. Soc., 1975, 97, 3039 CrossRef CAS.
- P. K. Pal, M. G. B. Drew and D. Datta, manuscript in preparation.
- I. Butala and G. Karlovic, Liebigs Ann. Chem., 1976, 1455 Search PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1988, 21, 916 CrossRef CAS.
- G. M. Sheldrick, Acta Cystallogr., Sect. A, 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-93, A Program for Crystal Structure Solution and Refinement, University of Göttingen, Germany, 1993..
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2002 |