Heterogeneous enantioselective hydrogenation of 2-pyrones over cinchona-modified palladium
Wolf-Rüdiger
Huck
,
Tamas
Mallat
and
Alfons
Baiker
*
Laboratory of Technical Chemistry, Swiss Federal Institute of Technology, ETH Hönggerberg-HCI, CH-8093 Zürich, Switzerland. E-mail: baiker@tech.chem.ethz.ch; Fax: +41 1 632 11 63; Tel: +41 1 632 31 53
First published on 7th January 2002
Abstract
Asymmetric hydrogenation of 4-alkoxy and 4-methyl derivatives of 2-pyrones to the corresponding dihydro- or tetrahydropyrones over cinchona-modified Pd/TiO2 is fast under ambient conditions and affords good yields and high ee. cis-Tetrahydropyrones are obtained with 98–99% de. The ee can be further increased by kinetic resolution.
The partial hydrogenation products of 2-pyrones are frequent patterns in natural products and have attracted considerable interest from the pharmaceutical industry due to their sedative, anti-convulsive, anaesthetic and antifungal properties. Some known example are dihydrokawain 5b and dihydromethysticin 6b (Scheme 1).1 Hydrogenation of substituted 2-pyrones with a chiral Ru complex catalyst afforded up to 98% ee and good chemoselectivity. A drawback of this homogeneous catalyst is that tetrahydropyrones are produced only as mixtures of cis and trans diastereoisomers with up to 80–90% de.2
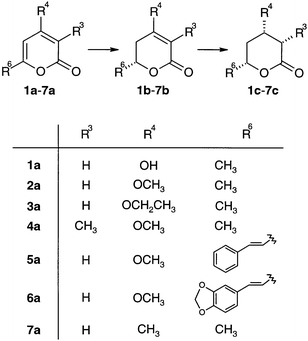 | ||
| Scheme 1 Hydrogenation of 2-pyrone derivatives over cinchonine-modified Pd/TiO2. | ||
Intrigued by the importance of chiral pyrones, we have attempted to carry out the hydrogenation reaction with chirally-modified metal catalysts. At best, a cinchona-modified Pd/TiO2 catalyst afforded 85% ee in the partial hydrogenation of 4-hydroxy-6-methyl-2-pyrone 1a to the corresponding 5,6-dihydropyrone 1b in acetonitrile.3 Though the transformation represents the first successful enantioselective hydrogenation of a semi-aromatic compound over a heterogeneous catalyst, the synthetic potential of the reaction is limited by the low reaction rate. Enantiodifferentiation has been assumed to be due to the formation of a bidentate complex between reactant and modifier, in which the interaction of the acidic hydroxy group of 1a with the quinuclidine N of cinchonidine plays a crucial role.4
Here, we report that not only 4-hydroxy, but also 4-alkoxy and 4-methyl derivatives of 2-pyrone, can be hydrogenated with high ee and good yield. This finding indicates that the asymmetric hydrogenation of 2-pyrone derivatives has a much broader scope than initially expected from the reaction mechanism proposed for 4-hydroxy-6-methyl-2-pyrone.4
The enantioselective hydrogenation of 4-alkoxy and 4-methyl derivatives of 2-pyrone was fast, even under ambient conditions, in various polar solvents, such as alcohols, THF or DMF. The highest yield and ee were mostly achieved in iPrOH, and these results are shown in Table 1. Under standard conditions and in the presence of cinchonine as chiral modifier, hydrogenation of 2a afforded 2b with 87% ee (entry 1). At room temperature and under atmospheric pressure the conversion was 93% and the chemoselectivity 80% after 2 h. The higher chemoselectivity of 1b (up to 90%) is due to tautomeric stabilization. Further hydrogenation of 2b was highly selective to the corresponding cis-2c, affording 99% de. Over-hydrogenation could be diminished by increasing the solvent volume (entry 2). Parallel to an increase of chemoselectivity from 80 to 90%, the ee increased to 90%. No further enhancement in ee and chemoselectivity was observed when using greater amounts of iPrOH, but the ee could be improved up to 94% by decreasing the amount of reactant (entry 3). Note that in the latter case, the reactant : surface Pd atom molar ratio was only 4 : 1, compared to 100 : 1 under standard conditions. For comparison, the homogeneous chiral Ru complex afforded the highest ee at a reactant/Ru molar ratio of 25.5
| Product b | Product c | ||||||
|---|---|---|---|---|---|---|---|
| Entry | Reactant | Time/h | Yield (%) | ee (%) | Yield (%) | ee (%) | de (%) |
a Conditions: 25 mg reactant, 20 mg 5 wt% Pd/TiO2, 15 ml iPrOH, 1 mg cinchonine, 1 bar, 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. n.d.=not determined.
b 50 ml iPrOH.
c 50 ml iPrOH, 1 mg reactant.
d 15 ml DMF. °C. n.d.=not determined.
b 50 ml iPrOH.
c 50 ml iPrOH, 1 mg reactant.
d 15 ml DMF.
|
|||||||
| 1 | 2a | 2 | 74 | 87 (R) | 19 | 86 (4S,6R) | 99 |
| 2 | 2a b | 2 | 80 | 90 (R) | 9 | 89 (4S,6R) | 98 |
| 3 | 2a c | 0.5 | 76 | 94 (R) | 4 | n.d. | n.d. |
| 4 | 3a | 2 | 71 | 85 (R) | 10 | 84 (4S,6R) | 99 |
| 5 | 4a | 2 | 8 | 75 (R) | 0 | — | — |
| 6 | 5a | 2 | 60 | 82 (R) | 29 | 80 (4S,6R) | 99 |
| 7 | 5a c | 1 | 67 | 89 (R) | 27 | 88 (4S,6R) | 99 |
| 8 | 6a | 2 | 68 | 82±2 (R) |
13 | n.d. | 99 |
| 9 | 6a c | 1 | 75 | 90±2 (R) |
18 | n.d. | 99 |
| 10 | 7a | 0.6 | 0 | — | 100 | 63 (4S,6R) | 99 |
| 11 | 7a d | 1 | 0 | — | 100 | 75 (4S,6R) | 99 |
Replacement of the 4-methoxy function by an ethoxy group (3a) had only a minor effect on the reaction rate and selectivities. In contrast, an additional methyl group in the α-position to the carbonyl group (4a) resulted in a dramatic drop in reactivity and the lowest ee (75%) in the series of 2-pyrones investigated here.
The influence of a bulky R6 alkyl group was studied with the reactants dehydrokawain 5a and dehydromethysticin 6a. In both reactions the hydrogenation proceeded stepwise. As expected on the basis of the general selectivity pattern of Pd,6 the alkene C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C bond in R6 was saturated first before the pyrone structure was hydrogenated (higher than 97% chemoselectivity after ca. 5 min). The yields to 5b and 6b and the ee were only moderately lower, compared to those for the hydrogenation of 2a
(cf. entries 1, 6, 8). The chemoselectivity could be increased up to 81% at 93% conversion of methysticin by dilution of the reaction mixture (entry 9). At almost full conversion (99%, 3 h), slow hydrogenolysis of the C–OCH3 bond was observed.
C bond in R6 was saturated first before the pyrone structure was hydrogenated (higher than 97% chemoselectivity after ca. 5 min). The yields to 5b and 6b and the ee were only moderately lower, compared to those for the hydrogenation of 2a
(cf. entries 1, 6, 8). The chemoselectivity could be increased up to 81% at 93% conversion of methysticin by dilution of the reaction mixture (entry 9). At almost full conversion (99%, 3 h), slow hydrogenolysis of the C–OCH3 bond was observed.
The potential of the Pd–cinchona system is not limited to 4-hydroxy- and 4-alkoxy-pyrones, that is, to the presence of an O-containing functional group in the 4-position, which may interact with the chiral modifier. The 4-methyl homolog 7a was rapidly hydrogenated to cis-7c with 99% de, and 63 or 75% ee in iPrOH and DMF, respectively (entries 10, 11). Due to the missing stabilization effect of the alkoxy function, separation between the uptake of two equivalents of hydrogen was poor, as illustrated in Fig. 1(a). The maximal yield of 7b was only 28% at 59% conversion of 7a. The ee in the hydrogenation of 7b to cis-7c was modified by a kinetic resolution [Fig. 1(b)]. The initial ee of 7b increased from 78 to 84%
(R) due to faster
hydrogenation of (S)-7b. Consequently, the ee of cis-7c increased from 56 (4S,6R) to 72%. The cumulative ee for both hydrogenation products was calculated to be constant during the whole reaction (72±1%).
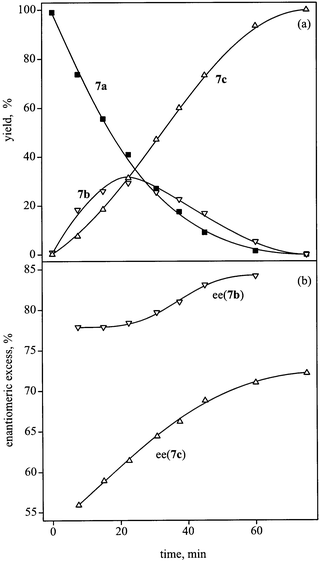 | ||
| Fig. 1 (a) Formation of dihydro- (7b) and tetrahydropyrone (7c) during reduction of 7a; composition of the reaction mixture (yield)
versus time. (b) Kinetic resolution of dihydropyrone (7b). Conditions: 50 mg 7a, 20 mg 5 wt% Pd/TiO2, 15 ml DMF, 1 mg cinchonine, 1 bar, 26°C. | ||
All tetrahydropyrones were obtained with 98–99% de. Using cinchonidine instead of cinchonine as the modifier, the ee were lower by 3–8% compared to entries 1–4 and 10 in Table 1. The effect of kinetic resolution was more pronounced by a factor of 2–3 when cinchonidine was applied instead of cinchonine with all the 2-pyrone derivatives tested, introducing the possibility of further enhancement in ee of the dihydropyrone at the expense of reducing the chemoselectivity. Note that the tetrahydropyrones can easily be removed by re-crystallization from hexane.
Pressures higher than 1 bar enhanced the rate, but lowered the ee. The reaction rate also increased with increasing temperature but the best enantioselectivities were achieved at 25–27°C. In this limited parameter study, only one type of catalyst was involved (5 wt% Pd/TiO2).
The good stereoselectivity achieved with 7a indicates that an O-containing functional group at C4 is not an essential requirement for enantiodifferentiation in the enantioselective hydrogenation of 2-pyrones, as may be inferred from a previous study of the hydrogenation of 4-hydroxy-6-methyl-2-pyrone.4 It seems that interaction of the cinchona alkaloid with the ester function of 7a on the Pd surface is sufficient. The observation that the size of the R6 alkyl groups has no significant influence on the ee allows additional variations in the substrate structure. Further advantages of the method are that both enantiomers can be obtained with similar ee's by using the cheap and easily available cinchonidine or cinchonine as chiral modifiers, and the possibility of improving the ee by kinetic resolution.
A comparison of our results in the partial hydrogenation of 4-alkoxy-2-pyrones with those reported for a chiral Ru complex reveals that cinchona-modified Pd/TiO2 offers a real alternative to the homogeneous catalyst—a rare situation in asymmetric catalysis. Pd provides comparable, and in some cases even better, enantioselectivities than the Ru complex. A characteristic feature of supported Pd is the high cis selectivity in olefin hydrogenation, affording 98–99% de in all reactions studied (Table 1). In some cases, the chemoselectivity to the partially hydrogenated product is also better with the Pd–cinchona system. For example, 6b was obtained in 75% yield (Table 1), compared to 27%, even at a low substrate/Ru catalyst ratio of 25.6
The asymmetric hydrogenation of alkyl- and alkoxy-substituted 2-pyrones reported in this work represents a significant extension of the narrow scope of CC bond hydrogenation over chirally-modified palladium.7 The enantioselectivity achieved in this work is the highest ever reported for heterogeneous asymmetric hydrogenation of CC bonds over chirally modified metals.
Further improvements in yields and selectivities seem achievable by optimizing the structural and chemical properties of the supported palladium catalyst.
Experimental
Cinchonine and 2-propanol (solvent) were used as supplied (Fluka). DMF was distilled under reduced pressure before use. 7a (Aldrich) was sublimated in vacuum and re-crystallized from hexane. 2a,83a,84a,8,95a10 and 6a10 were synthesized as described, sublimated or purified by flash chromatography, and re-crystallized from hexane. A 5 wt% Pd/TiO2 (metal dispersion: 18%, determined by H2 chemisorption) was prepared by precipitation of Pd hydroxide onto suspended TiO2.In a standard procedure, 20 mg catalyst was pre-reduced by H2 for 5 min, at 1 bar and 26°C in 13 ml solvent. Then 1 mg cinchonine in 1 ml solvent was added and after 5 min stirring the reaction was started by addition of the appropriate amount of reactant in 1 ml solvent. Conversion and chemo-, enantio- and diastereoselectivity were directly determined by an HP 6890 gas chromatograph. Enantioselectivities of 2b, 3b, 5b, 5c, 6b and 7c were analyzed with a Chirasil-DEX CB column (Chrompack) and 2c, 3c, 4b and 7b with a CyclosilB column (J+W). The integral ee of 7b and 7c was calculated as Σee=[ee(7b)·yield(7b)+ee(7c)·yield(7c)]/[yield(7b)+yield(7c)].
The (S)-enantiomer of all of the dihydropyrones eluted first, in agreement with the literature.2 The configuration of the tetrahydropyrones was deduced from these data by excluding complete inversion of ee in the second hydrogenation step, as supported by the conversion dependence of the ee of these products.
Acknowledgements
Financial support by the Swiss National Science Foundation is kindly acknowledged.References
- C. Spino, N. Mayes and H. Desfossés, Tetrahedron Lett., 1996, 37, 6503 CrossRef CAS.
- M. J. Fehr, G. Consiglio, M. Scalone and R. Schmid, J. Org. Chem., 1999, 64, 5768 CrossRef CAS.
- W.-R. Huck, T. Mallat and A. Baiker, J. Catal., 2000, 193, 1 CrossRef CAS.
- W.-R. Huck, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 2001, 200, 171 CrossRef CAS.
- M. J. Fehr, PhD Thesis no. 12789, Swiss Federal Institute of Technology, Zürich, 1998..
- G. V. Smith and F. Notheisz, Heterogeneous Catalysis in Organic Chemistry, Academic Press, San Diego, 1999. Search PubMed.
- T. Mallat and A. Baiker, in Fine Chemicals through Heterogeneous Catalysis, ed. R. A. Sheldon, H. van Bekkum, Wiley-VCH, Weinheim, 2001, p. 449. Search PubMed.
- P. De March, M. Morena-Mañas, R. Pi, I. Ripoll and F. Sánchez-Ferrando, J. Heterocycl. Chem., 1985, 22, 1537 Search PubMed.
- E. Ziegler, O. S. Wolfbeis and I. Trummer, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1982, 37, 105 Search PubMed.
- W. Adam, C. R. Saha-Möller, M. Veit and B. Welke, Synthesis, 1994, 1133. Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2002 |