(Ferrocenylmethyl)trimethylammonium cation: a very simple probe for the electro-chemical sensing of dihydrogen phosphate and ATP anions†
Olivier
Reynes
a,
Jean-Claude
Moutet
*a,
Jacques
Pecaut
b,
Guy
Royal
a and
Eric
Saint-Aman
a
aLaboratoire d'Electrochimie Organique et de Photochimie Rédox (CNRS UMR 5630), Université Joseph Fourier, BP 53, 38041 Grenoble cedex 9, France. E-mail: Jean-Claude.Moutet@ujf-grenoble.fr
bLaboratoire de Chimie de Coordination, DRFMC∣CEA∣SCIB, 17 rue des Martyrs, 38054 Grenoble cedex 9, France
First published on 10th January 2002
Abstract
By virtue of strong ion-pairing interactions that are reinforced following its oxidation to the ferrocinium form, (ferrocenylmethyl)trimethylammonium cation is able to electrochemically sense dihydrogen phosphate and ATP anions in organic electrolytes; clear two-wave voltammetry features allow their amperometric titration by this very simple derivative of ferrocene.
Anion binding plays a central role in chemical and biochemical processes and their recognition by artificial molecular hosts is an area of intense current interest.1 An application of anion receptor chemistry would be the qualitative and quantitative sensing of selectively bound anionic species. To this end, considerable attention has focused on the incorporation of a redox probe into a host structure to enable the detection of guest species through the perturbation of the redox system provoked by host–guest interactions.2 Of the redox centres used, the most prevalent have been the metallocenes, mainly cobaltocenium and ferrocene moities.2 Receptors containing neutral ferrocenyl units exhibit interesting electrochemical anion recognition effects, because electrostatic interactions can be switched on by oxidation of ferrocene to its cationic ferrocinium form. Ferrocene-based hosts can co-ordinate and electrochemically recognize anionic guest species via the cooperative binding forces of electrostatic interaction and other favourable interactions such as hydrogen bonding with appended thiourea,3 guanidinium,3 amide4–7 or pyrrole8 H-donor groups. Additional amine,5 pyridine6 or polypyridine9 H-bond acceptor groups can enhance the interaction between the receptor and oxoanions such as dihydrogen phosphate and hydrogen sulfate anions. Shape selectivity and topological effects in hosts based on macrocyclic calixarene10 and cyclotriveratrylene11 platforms or in dendrimers12 and colloids13 also influence both anion–receptor interactions and guest recognition. However, basic ion pairing appears the most important feature to be exploited in the construction of redox-active species able to electrochemically recognize anions, especially in non-aqueous media. We herein report the remarkable electrochemical sensing properties of the ordinary (ferrocenemethyl)trimethylammonium cation 1 towards dihydrogen phosphate and adenosine-5′-triphosphate anions (ATP2−) in various organic electrolytes.
The cyclic voltammetry (CV) curves for 1 exhibit the regular wave corresponding to the reversible ferrocene/ferrocinium (Fc/Fc+) redox couple in acetonitrile, dichloromethane or acetone containing 0.1 M tetra-n-butylammonium perchlorate (TBAP). Due to different solvation effects, E1/2 ranges from 0.17 to 0.22 V, depending on the solvent used (Table 1). Progressive addition of hydrogen sulfate and nitrate produced weak negative shifts of the Fc/Fc+ oxidation wave (Table 1). With nitrate, the maximum perturbation of the CV curve (−10 mV) was obtained with 1 equiv. of added anion. Larger shifts of the Fc/Fc+ wave (up to −100 mV in CH2Cl2) were observed upon addition of an excess of hydrogen sulfate anion (Table 1). Moreover, due to the weak solubility of ion pairs formed between the oxidized dicationic probe and HSO4− anions, their dissolution upon reduction are responsible for intense stripping peaks in the presence of a large excess of anion, especially in the less polar acetone and dichloromethane solvents. With F− the electrochemical response of 1 is characterized by a large increase in the anodic peak current, along with a decrease in the reversibility of the Fc/Fc+ wave, which progressively shifts towards more negative potentials. This behaviour is characteristic of an EC mechanism with product adsorption and suggests that strong ion pairs formed between oxidized 1 and F− remain strongly adsorbed onto the electrode surface. Increasing amounts of Cl−, Br− or SCN− do not produce any significant change in the CV wave although, as for other ferrocenoyl compounds,9 catalytic oxidation of Cl− clearly occurs upon addition of more than 4 equiv. of chloride and the ferrocene groups partially decompose to form [FeClx] species.
| Solvent | Free 1E1/2/V |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +H2PO4−
ΔEb/mV +H2PO4−
ΔEb/mV |
1+ATP2−
ΔEb/mV |
1+HSO4−ΔEc/mV |
1+NO3−
ΔEc/mV |
|---|---|---|---|---|---|
|
a
Versus ferrocene; the different anions were used as their tetra-n-butylammonium salts; ν=0.1 V s−1; E1/2=(EPa+EPc)/2; EPa and EPc are the oxidation and reduction peak potentials, respectively; ΔE=EPa
([A−]≠0)−EPa
([A−]=0).
b “Two-wave” behaviour.
c “One-wave” behaviour.
d Measured in the presence of 4 equiv. of HSO4−.
e Not determined.
f No recognition.
g Measured in the presence of 1.5 equiv. of ATP2−.
|
|||||
| CH3CN | 0.28 | −295 | −235 | −10 (−15d) | −10 |
| CH2Cl2 | 0.27 | −470 | −280 | −25 (−100d) | −10 |
| (CH3)2CO | 0.29 | −320 | —e | −10 (−35d) | −10 |
| CH3OH | 0.34 | —f | −30cg | —f | —f |
In contrast, a remarkable two-wave behaviour was obtained when considering the 1+H2PO4− system. Fig. 1(A) shows the changes in the CV wave of 1 in the presence of increasing amounts of dihydrogen phosphate in CH2Cl2+TBAP electrolyte. Successive additions of this anion result in the extinction of the initial Fc/Fc+ wave and the appearance of a new wave corresponding to complexed redox probe. The new wave grows at less positive potentials than the original Fc/Fc+ wave (−295 to −470 mV, depending on the solvent used; see Table 1). Maximum perturbation of the CV curves is obtained with 3–4 equiv. of added H2PO4− anion. This clear electrochemical behaviour allows an amperometric titration
curve to be drawn by considering the intensity of the new anodic peak Ipacvs. the H2PO4−/1 molar ratio [Fig. 1(B)]. Ipac increases linearly with the amount of anion added to the solution, until reaching a maximum. The slow decrease in current at higher concentrations of anion is due to some precipitation of the 1+H2PO4− complex.
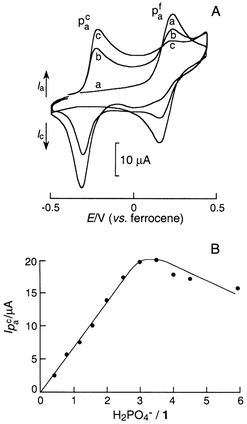 | ||
| Fig. 1
(A) Cyclic voltammograms, recorded at a Pt disc electrode (5 mm in diameter) in CH2Cl2+0.1 M TBAP, of 0.6 mM 1: (a) free 1; (b) H2PO4−/1=1.6; (c) H2PO4−/1=3. Sweep rate 0.1 V s−1. (B) Amperometric titration of dihydrogen phosphate in CH2Cl2: increase in the intensity of the new wave vs. the number of equivalents of H2PO4− added per (ferrocenylmethyl)trimethylammonium. | ||
The behaviour of the 1+H2PO4− system in acetone is close to that observed in dichloromethane. In CH3CN, strong adsorption-desorption phenomena are responsible for a larger peak-to-peak separation and for the appearance of a sharp stripping peak in the reverse scan. A similar behaviour was observed with the ATP2− anion in CH3CN and CH2Cl2
(Table 1). However, no reduction peak was seen after the addition of 1 molar equivalent of anion, due to the very poor solubility of the ion pairs formed between the oxidized 1 and ATP2−.
Compound 1 is thus potentially able to sense anionic guest species via electrostatic interaction with the appended quaternary ammonium group, which is further reinforced following oxidation to ferrocinium. The binding of anions effectively stabilizes the double positive charge of the oxidized form of 1 causing the Fc/Fc+ redox couple to shift to less positive potentials. As shown in Table 1, polarity of the solvent plays a central role in the recognition ability of 1. Potential shifts are significantly higher in CH2Cl2 than in CH3CN, due to an increase in the electrostatic interaction in the less polar solvent. Solvation can, however, counteract ion-pair formation. This was evidenced by studies carried out in methanol, which presents a dielectric constant close to that of acetonitrile. In this protic solvent stabilization of anions due to hydrogen-bonding
interactions is responsible for a complete loss of electrochemical sensing ability of 1 towards almost all the surveyed anions, with the exception of ATP2−, which induces a small negative shift (ΔE=−30 mV with 1.5 equiv. of anion) in the potential of the Fc/Fc+ redox wave (Table 1).
In considering the observed selective sensing response one should be aware of the interaction between neutral/oxidized 1 and the different anions. The association constants Ka were determined for 1 with HSO4−, NO3− and F− by a standard 1H-NMR titration,11 monitoring Δδ(CH2N+) and Δδ[(CH3)3N+] in CD3CN, CD2Cl2 and CD3COCD3+0.1 M TBAP solutions of 1
(10−2 M) with the addition of increasing amounts of a given anion (Table 2). In CD3CN we found weak, similar binding constants with the hydrogen sulfate, nitrate and fluoride anions (Ka=11,
9 and 10 M−1, respectively). The interaction of ATP2− with 1 is significantly stronger (Ka=122 M−1), in agreement with the better electrochemical sensing properties of 1 towards this anion (Table 1). It is noteworthy that the association constants are significantly higher in CD2Cl2 and CD3COCD3, due to an increase in electrostatic interactions in these less polar solvents. Unfortunately, in all solvents precipitation of 1+H2PO4− ion pairs onsets when more than 1 equiv. of H2PO4− is added and the association constants could not be determined due to these constraints. However, the large ΔE values measured in the different electrolytes (Table
1) mean that the apparent association constants between H2PO4− or ATP2− and the oxidized form of 1 are several orders of magnitude larger than the association constants with reduced 1,12,14 following the establishment of very strong electrostatic interactions between the ferrocinium form of 1 and these anions.15
| Solvent | F− | HSO4− | NO3− | ATP2− |
|---|---|---|---|---|
|
a Determined from 1H-NMR data (see the text) at T=294 K; in all solvents precipitation of 1+H2PO4− salt precluded determination of Ka.
b Not determined, due to the poor solubility of the anion.
|
||||
| CD3CN | 10.0±0.8 |
11.0±1.2 |
9.0±1.7 |
122±6 |
| CD2Cl2 | —b | 61±10 |
—b | 185±12 |
| (CD3)2CO | —b | 38±2 |
—b | 159±18 |
The 1·H2PO4− complex was isolated in the solid state (see Experimental). ES mass spectral analysis confirmed the formation of strong ion pairs between 1 and H2PO4− and gave information on the stoichiometry of the species formed. The mass spectrometric study clearly showed the presence in dry acetonitrile of five different ion pairs: [1(H2PO4−)2]−, [(1)2(H2PO4−)3]−, [(1)2(H2PO4−)4]2−, [(1)3(H2PO4−)4]− and [(1)3(H2PO4−)5]2−.
This result is in keeping with the poorly defined stoichiometry (1+3–4 H2PO4−) determined from electrochemical data.
X-Ray quality crystals were grown at 5°C by slow diffusion of diethyloxide in an ethanol solution of 1·H2PO4−. The X-ray structures revealed (Fig. 2) that the crystals of 1·H2PO4− belong to the P2(1)/c space group of the monoclinic system. 1·H2PO4− crystallizes in a channel structure (Fig. 2). The ammonium group of 1 points towards the H2PO4− anion, each H2PO4− anion being surrounded by four (ferrocenemethyl)trimethylammonium cations with P−N distances ranging from 4.708 to 5.040 Å and P–O distances ranging from 1.498 to 1.565 Å. The P–P distance between two closest
H2PO4− anions in a channel is ca. 4.30 Å. Both Cp rings are almost coplanar with a small Cp–Fe–Cp bent angle of 2.4° and the intramolecular distances (3.28 Å) in the Cp rings are as expected for a substituted ferrocene. One water and one ethanol molecule are located in the unit cell and these solvent molecules are strongly hydrogen-bonded to the H2PO4− anion with an H⋯A distance between 1.93(2) and 2.07(3)
Å. An H-bonding interaction is revealed between a hydrogen atom from a methyl group in the ammonium headgroup of 1 and an oxygen atom of H2PO4− with an H⋯O distance of 2.41(3)
Å, contributing to a reinforcement of the interactions between 1 and H2PO4− in addition to the electrostatic forces.
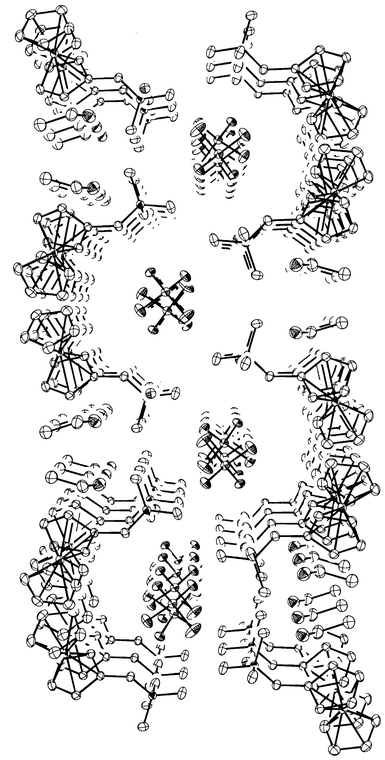 | ||
| Fig. 2 Packing view down the c axis showing the location of dihydrogen phosphate anions in channels formed by (ferrocenylmethyl)trimethylammonium cations and the presence of ethanol and water (located between ammonium headgroups) in the framework. H atoms are omitted for clarity. | ||
The selective sensing of the dihydrogen phosphate anion is believed to be due to specific electrostatic interactions with 1 and its oxidized form. In agreement with this statement, we found that addition of dihydrogen phosphate to an acetonitrile solution of unsubstituted ferrocene causes the development of a stripping peak on the reduction branch of the CV wave while no change occurred in the presence of hydrogen sulfate anions. Furthermore, an additional Lewis acid-base interaction with the iron centre of the ferrocene could also be involved. 1H-NMR experiments showed evidence for slight interactions between unsubstituted ferrocene and dihydrogen phosphate, and to a lesser extent hydrogen sulfate; the H-Cp resonances being shifted downfield by 0.2 ppm and 0.06 ppm upon addition of one equivalent of H2PO4− and HSO4−, respectively. This might be due to the greater basicity of the former anion.
The ferrocenetetraalkylammonium 1 is thus a very simple redox probe whose selective electrochemical sensing properties towards H2PO4− and ATP2− in organic media are mainly a direct consequence of a strong ion-pairing interaction. Both the specific Fc/Fc+ redox peak system and the large potential shifts found with these anions suggest the potential use of 1 and its derivatives in the construction of amperometric sensor devices. We are currently investigating the synthesis and the redox sensing properties of polymer films containing cationic derivatives of ferrocene.
Experimental
Electrochemical experiments were conducted in a three-electrode cell under an argon atmosphere and at room temperature, with as working electrode a platinum or a carbon disc (5 and 3 mm in diameter respectively). Potential data are referenced to the potential of the unsubstituted ferrocene/ferrocinium redox couple. Acetonitrile (Rathburn, HPLC grade S), acetone and methanol (analytical reagent grade) were used as received. Dichloromethane (analytical reagent grade) was dried over neutral aluminium oxide (activity I) for a least 4 days before use. Tetra-n-butylammonium perchlorate (Fluka) was dried under vacuum at 80°C for 3 days. Tetraethylammonium fluoride and nitrate, tetra-n-butylammonium hydrogen sulfate and dihydrogen phosphate were of the highest purity commercially available and were used without further purification. The di(tetra-n-butylammonium)
salt of adenosine-5′-triphosphate (ATP2−) was obtained from its corresponding disodium salt by ion exchange on Amberlite IRC50 in n-Bu4N+ form.
(Ferrocenylmethyl)trimethylammonium iodide16 was converted to its corresponding tetrafluoroborate salt by metathesis with NH4BF4. The mixture was extracted with CH2Cl2 and the orange extract was dried over Na2SO4. Addition of Et2O gave 1·BF4− as a dark-yellow powder; yield 60%. Analytical data: 1H-NMR (250 MHz, CD3CN, 10 mM, 295 K) δ: 2.86 [s, 9H, N+(CH3)3], 4.23 (s, 5H, Cp-H), 4.28 (s, 2H, Cp–CH2–N+), 4.38 (m, 2H, Cp-Hβ), 4.44 (m, 2H, Cp-Hα); 13C-NMR (250 MHz, CD3CN, 10 mM, 295 K) δ: 52.79 [N+(CH3)3], 67.76 (Cp–CH2–N+), 70.14, 71.42, 73.08, 73.47 (Cp); UV-vis (CH3CN) λmax/nm (ε/M−1 cm−1): 431 (75), 335 (30); FAB+-MS: m/z 258 (1+).
1·H2PO4− was obtained by adding an excess of tetra-n-butylammonium dihydrogen phosphate (4 molar equivalents) to an acetonitrile solution of 1·BF4−
(5 mmol in 3 mL) at room temperature under stirring. A pale yellow precipitate was formed instantaneously. The solid was isolated by suction filtration and washed with cold acetonitrile. Analytical data: 1H-NMR (250 MHz, CD3OD, 10 mM, 295 K)
δ: 2.98 [s, 9H, N+(CH3)3], 4.26 (s, 5H, Cp-H), 4.42 (s, 2H, Cp–CH2–N+), 4.43 (m, 2H, Cp-Hβ), 4.49 (m, 2H, Cp-Hα); UV-vis (C2H5OH)
λmax/nm (ε/M−1
cm−1): 432 (63), 339 (22). Anal. calcd for C14H22FeNO4P·H2O·CH3OH (Mr=405.214): C, 44.46; H, 6.96; N, 3.46; P, 7.64; Fe, 13.78; found C, 44.99; H, 6.68; N, 3.59; P, 7.40; Fe, 13.18%.
X-Ray quality crystals were grown by vapour diffusion of Et2O into an ethanol or methanol solution of 1·H2PO4− at 5°C. Crystal data: 1·H2PO4−·H2O·C2H5OH: C14H22FeNO4P·H2O·C2H6O, Mr=419.23, crystal size=0.08×0.5×0.5 mm, a=17.024(5)
Å, α=90°, b=13.573(3)
Å, β=95.710(15)°, c=8.4498(19)
Å, γ=90°, U=1942.8(9)
Å3, T=223(2)
K, λ=0.71073 Å, monoclinic, space groups P2(1)/c, Dc=1.433 Mg m−3, μ=0.889 mm−1, collected reflections=12266, final R
[I>2σ(I)]=0.0301, wR=0.0739, R indices (all data)=0.0517, wR=0.0882. The data sets for the single-crystal X-ray study were collected with Mo-Kα radiation on a Brucker SMART diffractometer. All calculations were performed using the SHELXTL program.17 The structure was solved by direct methods and refined with full-matrix least-squares on F2.
CCDC reference number 176826. See http://www.rsc.org/suppdata/nj/b1/b107713a/ for crystallographic data in CIF or other electronic format.
1H-NMR experiments were conducted at 21°C on a Brucker AC250 spectrometer using the solvent deuterium signal as internal reference. ES mass spectra were recorded on a Micromass Quattro mass spectrometer. 1·H2PO4− was dissolved (0.5 mg ml−1) in dry CH3CN and samples were introduced into the ES source with a flow rate of 5 μl min−1.
References
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS.
- P. D. Beer and J. Cadman, Coord. Chem. Rev., 2000, 205, 131 CrossRef CAS.
- P. D. Beer, M. G. B. Drew and D. K. Smith, J. Organomet. Chem., 1997, 543, 259 CrossRef CAS.
- P. D. Beer, Z. Chen, A. J. Goulden, A. R. Graydon, S. E. Stokes and T. J. Wear, J. Chem. Soc., Chem. Commun., 1993, 1834 RSC.
- P. D. Beer, A. R. Graydon, A. O. M. Johnson and D. K. Smith, Inorg. Chem., 1997, 36, 2112 CrossRef CAS.
- J. D. Carr, L. Lambert, D. E. Hibbs, M. B. Hursthouse, K. M. A. Malik and J. H. R. Tucker, Chem. Commun., 1997, 1649 RSC.
- K. Kavallieratos, S. Hwang and R. H. Crabtree, Inorg. Chem., 1999, 38, 5184 CrossRef CAS.
- M. Sherer, J. L. Sessler, A. Gebauer and V. Lynch, Chem. Commun., 1998, 85 RSC.
- M. Buda, A. Ion, J.-C. Moutet, E. Saint-Aman and R. Ziessel, J. Electroanal. Chem., 1999, 469, 132 CrossRef CAS.
- P. A. Gale, Z. Chen, M. G. B. Drew, J. A. Heath and P. D. Beer, Polyhedron, 1998, 17, 405 CrossRef.
- O. Reynes, F. Maillard, J.-C. Moutet, G. Royal, E. Saint-Aman, G. Stanciu, J.-P. Dutasta, I. Gosse and J.-C. Mulatier, J. Organomet. Chem., 2001, 637–639, 356 CrossRef CAS.
- C. Valerio, J.-L. Fillaut, J. Ruiz, J. Guittard, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 1997, 119, 2588 CrossRef CAS.
- A. Labande and D. Astruc, Chem. Commun., 2000, 1007 RSC.
- S. R. Miller, D. A. Gustowski, Z. H. Chen, G. W. Gokel, L. Echegoyen and A. E. Kaifer, Anal. Chem., 1988, 60, 2021 CrossRef CAS.
- It is noticeable that in the presence of other bases at least as strong as H2PO4−, such as N(C2H5)3 or tetra-n-butylammonium hydroxyde (TBAOH), the reversible electrochemical response of 1 turns irreversible. The CV curve recorded in the presence of N(C2H5)3 displays the features of an electrocatalytic mechanism and the full irreversibility of 1/1+ in the presence of TBAOH is likely to be due to a fast and irreversible degradation of the electrogenerated 1+ in the presence of OH−. The same electrochemical behaviour was found for the simple, unsubstituted ferrocene
in the presence of N(C2H5)3 or TBAOH. Thus, the large ΔE measured for 1+H2PO4− cannot be due to deprotonation of the highly acidic CH2 group between ferrocene and the ammonium unit by the basic phosphate anion..
- J. K. Lindsay and C. R. Hauser, J. Org. Chem., 1957, 22, 355 CrossRef CAS.
- G. M. Sheldrick, SHELXTL v. 5.1. An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffraction Data, Siemens Analytical X-Ray Instruments, Madison, WI, 1990..
Footnote |
| † Electronic supplementary information (ESI) available: cyclic voltammograms of 1 in the presence of increasing amounts of ATP2− or HSO4−. See http://www.rsc.org/suppdata/nj/b1/b107713a/ |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2002 |