A novel ligand family based on bulky metallocalix[4 and 8]arene substituents†
Vernon C.
Gibson
a,
Carl
Redshaw
*b and
Mark R. J.
Elsegood
c
aDepartment of Chemistry, Imperial College, South Kensington, London, UK SW7 2AY
bWolfson Materials and Catalysis Centre, School of Chemical Sciences, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: carl.redshaw@UEA.ac.uk
cChemistry Department, Loughborough University, Loughborough, Leicestershire, UK LE11 3TU
First published on 20th December 2001
Abstract
Condensation reactions of metallocalix[4 and 8]arene complexes containing pendant amino groups readily afford new bulky imine complexes; the X-ray crystal structures of three complexes, of which two were determined using synchrotron radiation, are reported.
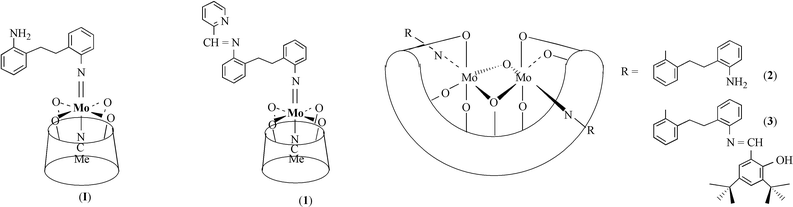
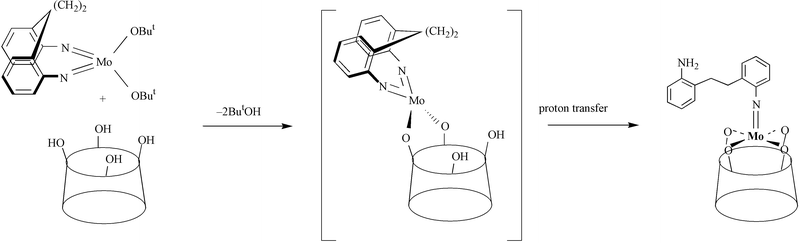
In recent years, there has been considerable and growing interest in new metallocalixarenes and the coordination chemistry thereof.1 Recent advances by Floriani and coworkers2 have utilised the simplest of the calixarenes, the calix[4]arene system (L1H4), particularly for its ability to act as a quasi-oxo support. The majority of these metallocalix[4]arene derivatives exist as either mono or binuclear complexes, retaining a cone-like conformation for the ligand. On the other hand, knowledge about the structure and reactivity of metal compounds containing the larger ring systems is comparatively scant.3 In particular, clear methodologies for studying the coordination chemistry of these larger ring systems remain in the early stages of development. The first organoimido metal calixarenes [Mo(NAr)L1(NCMe)] and [{Mo(NAr)(NCMe)}2L2] (Ar = C6H3Pri2-2,6; L2 = calix[8]arene or p-tert-butylcalix[8]arene), both of which were formed readily via alkoxide and imide displacement reactions, have been reported.3f This methodology was extended to include the related ansa-bis(imido) derivative {Mo(OBut)2[(2-NC6H4)2CH2CH2]}4 which may be ring-opened on reaction with L1H45 to release a pendant amino group (see 1). Such reactivity provided an opportunity to develop an entry into a new family of ligands bearing bulky metallocalixarene substituents, the first example of which are described here. These include the ready synthesis of bulky imino-metallocalix[4]arene complex [Mo(NAr1)L1] (2) (Ar1 = 2-C6H4CH2CH2C6H4N-2′-CHC5H5N). For the larger ring systems, we describe the preparation and structure of the dimetallocalix[8]arene complex {[Mo(NAr2)]2L2}·6MeCN (3) (L2 = p-tert-butylcalix[8]arene; Ar2 = 2-C6H4CH2CH2C6H4NH2-2′), containing two pendant amino groups, and its reactivity towards 3,5-di-tert-butylsalicylaldehyde at elevated temperatures to afford the bis(salicylaldiminato) complex {[Mo(NAr3)]2L2}·5MeCN (4) (Ar3 = 2-C6H4CH2CH2C6H4N-2′-CHC6H2-2′′-(OH)-3′′,5′′-But2), see Scheme 1. Furthermore, these Mo
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R–NH2 systems (R = Ar1, Ar3, etc.) have potential for the synthesis of unusual amines via cleavage of the remaining MoN bond (i.e. Mo–calixarene-mediated amine synthesis). Interestingly, novel salen-type calix[4]arene ligands (upper-rim) have recently been reported and their potential as rhenium-based radiopharmaceuticals
has been demonstrated.6
N–R–NH2 systems (R = Ar1, Ar3, etc.) have potential for the synthesis of unusual amines via cleavage of the remaining MoN bond (i.e. Mo–calixarene-mediated amine synthesis). Interestingly, novel salen-type calix[4]arene ligands (upper-rim) have recently been reported and their potential as rhenium-based radiopharmaceuticals
has been demonstrated.6
 | ||
| Scheme 1 | ||
The reaction of 1 with 2-pyridinecarboxaldehyde in refluxing ethanol affords, after work-up, the pyridyl-imino complex [Mo(NAr1)L1] (2) in good yield. The room temperature solution 1H NMR spectrum is consistent with the C4v-symmetric cone conformation. Crystals obtained from a saturated solution of acetonitrile at ambient temperature were too weakly diffracting to give reasonable structural details using a conventional sealed-tube X-ray source, but a data set was readily obtained using synchrotron radiation.7 The molecular structure is shown in Fig. 1 and selected bond lengths and angles are given in the caption. There are 2 uncoordinated molecules of solvent (MeCN) per molecule of the complex. The molybdenum centre possesses a pseudo-octahedral geometry similar to that found in the amino analogue 1, with the molybdenum atom displaced from the O4 mean plane towards the imido nitrogen N(1) by 0.245 Å.
![The molecular structure of 2. H-atoms, calix[4]arene p-But groups and 2 MeCN molecules of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Mo(1)–O(1) 1.931(7), Mo(1)–O(2) 1.940(7), Mo(1)–O(3) 1.942(7), Mo(1)–O(4) 1.929(7), Mo(1)–N(1) 1.738(7), Mo(1)–N(4) 2.324(7), N(1)–C(45) 1.361(11); Mo(1)–N(1)–C(45) 171.9(7).](/image/article/2002/NJ/b108088c/b108088c-f1.gif) | ||
| Fig. 1 The molecular structure of 2. H-atoms, calix[4]arene p-But groups and 2 MeCN molecules of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Mo(1)–O(1) 1.931(7), Mo(1)–O(2) 1.940(7), Mo(1)–O(3) 1.942(7), Mo(1)–O(4) 1.929(7), Mo(1)–N(1) 1.738(7), Mo(1)–N(4) 2.324(7), N(1)–C(45) 1.361(11); Mo(1)–N(1)–C(45) 171.9(7). | ||
In order to extend this approach to the larger p-tert-butylcalix[8]arene (L2) ring system, {Mo(OBut)2[(2-NC6H4)2CH2CH2]}4 was treated with H8L2 (0.5 equiv.) in toluene affording, after work-up, the bis(imido) complex {[Mo(NAr′)]2L2} (3), in which each imido ligand contains a pendant amino group. The IR spectrum of 3 contains two broad (weak) stretches at ca. 3365 and 3180 cm−1 in the ν(N–H) region, with the lower stretching constant associated with the H-bonded N(2) amino group. Complex 3 is presumed to form via loss of four tert-butanol ligands (two from each molybdenum) followed by proton transfer to each of the imido ligands, to release two pendant amino groups in an analogous fashion to that observed for [{Mo(NAr)(NCMe)}2(calix[8]arene)].3f Crystals of 3 suitable for an X-ray analysis were grown from acetonitrile solution at room temperature in ca. 48% yield. The molecular structure is shown in Fig. 2, with selected bond lengths and angles given in the caption. Each molybdenum possesses a pseudo-octahedral geometry, resulting in what is best described as a central edge-shared bioctahedral arrangement, featuring asymmetric phenoxide bridges [Mo(2)–O(1) = 2.247(4) cf. Mo(2)–O(5) = 2.061(5) Å] which are coplanar with terminal organoimido ligands.
![The molecular structure of 3. Most H-atoms, calix[8]arene p-But groups and 6 MeCN molecules of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Mo(1)–O(1) 2.078(4), Mo(1)–O(2) 1.925(5), Mo(1)–O(3) 1.940(5), Mo(1)–O(4) 1.961(5), Mo(1)–O(5) 2.216(5), Mo(1)–N(1) 1.735(6), Mo(2)–O(1) 2.247(4), Mo(2)–O(5) 2.061(5), Mo(2)–O(6) 1.940(5), Mo(2)–O(7) 1.940(5), Mo(2)–O(8) 1.957(4), Mo(2)–N(3) 1.727(6),
N(1)–C(1) 1.385(9), N(3)–C(15) 1.400(9); Mo(1)–N(1)–C(1) 178.1(5), Mo(2)–N(3)–C(15) 178.7(6).](/image/article/2002/NJ/b108088c/b108088c-f2.gif) | ||
| Fig. 2 The molecular structure of 3. Most H-atoms, calix[8]arene p-But groups and 6 MeCN molecules of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Mo(1)–O(1) 2.078(4), Mo(1)–O(2) 1.925(5), Mo(1)–O(3) 1.940(5), Mo(1)–O(4) 1.961(5), Mo(1)–O(5) 2.216(5), Mo(1)–N(1) 1.735(6), Mo(2)–O(1) 2.247(4), Mo(2)–O(5) 2.061(5), Mo(2)–O(6) 1.940(5), Mo(2)–O(7) 1.940(5), Mo(2)–O(8) 1.957(4), Mo(2)–N(3) 1.727(6), N(1)–C(1) 1.385(9), N(3)–C(15) 1.400(9); Mo(1)–N(1)–C(1) 178.1(5), Mo(2)–N(3)–C(15) 178.7(6). | ||
The conformation of the calixarene ring is such that each organoimido group is encapsulated by 3 calixarene-phenolate subunits—a double cup. The pendant C6H4NH2 [N(4)] group of one of the imido groups shows some signs of disorder, though this could not be successfully modelled. In contrast, the N(2)-containing group of the other imido ligand shows no sign of disorder, due to the H-bonding locking in the pendant arm [O(4)–H(2A) = 2.25 Å; O(4)–H(2A)–N(2) = 142°]. There are 6 MeCN solvent molecules in the asymmetric unit, one of which is very diffuse (or partially present). However, there is no solvent within the double cup calixarene cavities.
The ring opening reaction (see Scheme 2 for an example) described above gives a new dimension to the chemistry of imido-metallocalix[8]arenes and offers an entry point into complexes functionalised with a free reactive group. For example, reaction (condensation) of 3 with two equivalents of 3,5-di-tert-butylsalicylaldehyde in refluxing ethanol readily affords the bis(salicylaldimine) complex {[Mo(NAr″)]2L}
(4)
(Ar″
= 2-NC6H4CH2CH2C6H4N-2-CHC6H2-2′-(OH)-3″,5″-But2) in good yield (60–70%). As expected, the IR spectrum contains stretches in the ν(OH) region (broad/weak)
together with a strong band in the ν(CN) region. Small needle-shaped crystals of 4 suitable for an X-ray determination using synchrotron radiation7 were grown from acetonitrile at 0 °C; they incorporate 5 molecules of solvent per molecule of the complex.
 | ||
| Scheme 2 | ||
The molecular structure is shown in Fig. 3 and reveals the way in which the macrocyclic ring twists to adopt an edge-shared bioctahedron about the molybdenum centres. This ‘pinched’ conformation is similar to that observed for Na{Butcalix[8]arene[Ti(OPri)]2}.3d The highly functionalised ‘organoimido’ (salicylaldimine) ligands are clearly acting as four-electron donors [Mo(1)–N(1) = 1.723(3) Å; Mo(1)–N(1)–C(89) = 179.0(3)°]; the bridging phenoxides are asymmetric, with those trans to the ‘organoimido’ groups being substantially longer [Mo(1)–O(5) = 2.243(2) cf. Mo(1)–O(1) = 2.065(2) Å], as observed in complex 2. Each salicylaldimine group exhibits some internal H-bonding.
![The molecular structure of 4. Most H-atoms, calix[8]arene p-But groups and 5 MeCN molecules of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Mo(1)–O(1) 2.065(2), Mo(1)–O(2) 1.919(2), Mo(1)–O(3) 1.931(2), Mo(1)–O(4) 1.931(3), Mo(1)–O(5) 2.243(2), Mo(1)–N(1) 1.723(3), Mo(2)–O(1) 2.241(2), Mo(2)–O(5) 2.045(2), Mo(2)–O(6) 1.916(2) [note that O(6) is trans to O(8), but masked by Mo(2) in the diagram], Mo(2)–O(7)
1.937(2), Mo(2)–O(8) 1.948(2), Mo(2)–N(3) 1.722(3), N(1)–C(89) 1.393(5), N(3)–C(118) 1.398(5); Mo(1)–N(1)–C(1) 179.0(3), Mo(2)–N(3)–C(15) 176.5(2).](/image/article/2002/NJ/b108088c/b108088c-f3.gif) | ||
| Fig. 3 The molecular structure of 4. Most H-atoms, calix[8]arene p-But groups and 5 MeCN molecules of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Mo(1)–O(1) 2.065(2), Mo(1)–O(2) 1.919(2), Mo(1)–O(3) 1.931(2), Mo(1)–O(4) 1.931(3), Mo(1)–O(5) 2.243(2), Mo(1)–N(1) 1.723(3), Mo(2)–O(1) 2.241(2), Mo(2)–O(5) 2.045(2), Mo(2)–O(6) 1.916(2) [note that O(6) is trans to O(8), but masked by Mo(2) in the diagram], Mo(2)–O(7) 1.937(2), Mo(2)–O(8) 1.948(2), Mo(2)–N(3) 1.722(3), N(1)–C(89) 1.393(5), N(3)–C(118) 1.398(5); Mo(1)–N(1)–C(1) 179.0(3), Mo(2)–N(3)–C(15) 176.5(2). | ||
Future studies will be directed towards the use of these bulky ligands in transition metal chemistry.
Experimental
All manipulations were carried out under an atmosphere of nitrogen using standard Schlenk and cannula techniques, or in a conventional nitrogen-filled glove-box. Solvents were refluxed over an appropriate drying agent, and distilled and degassed prior to use.Selected data for 2–4
Elemental analysis for 2: calcd for C66H72O4N4Mo·0.5MeCN : C, 73.0; H, 6.7; N, 5.7; found: C, 73.3; H, 6.6; N, 5.9%. For 3: calcd for C116H132O8N4Mo2·6C2H3N: C, 71.5; H, 7.0; N, 6.5; found: C, 71.2; H, 6.9; N, 6.4%. For 4: calcd for C146H172O10N4Mo2·5C2H3N: C, 73.7; H, 7.4; N, 4.9; found: C, 73.5; H, 7.4; N, 4.6%.Selected spectroscopic data for 2: IR ν(CN) 1616 cm−1. 1H NMR (C6D6, 300 MHz, 298 K)
δ: 8.64 (s, 1H, CHN), 5.17 (d, 4H, 2JHH 12.2 Hz, endo-CH2), 4.33 (t, 2H, 2JHH 6.4 Hz, NCH2), 4.17 (t, 2H, 2JHH 7.1 Hz, NCH2), 3.55 (d, 4H, 2JHH 12.1 Hz, exo-CH2). For 3: IR: ν(NH2) 3365, 3181 cm−1; ν(C–N) 1263 cm−1. 1H NMR (C6D6, 300 MHz, 298 K)
δ: 5.42 (d, 2H, 2JHH 12.1 Hz, endo-CH2),
4.78 (d, 2H, 2JHH 13.9 Hz, endo-CH2), 4.54 (d, 2H, 2JHH 14.0 Hz, endo-CH2), 4.42 (d, 2H, 2JHH 13.9 Hz, endo-CH2), 3.25 (d, 2H, 2JHH 12.8 Hz, exo-CH2), 2.85 (d, 2H, 2JHH 12.7 Hz, exo-CH2) (2 ×
exo-CH2 obscured by overlap with ArCH2). For 4: IR ν(OH) 3370 cm−1, ν(CN) 1616 cm−1. 1H NMR (C6D6, 300 MHz, 298 K)
δ: 14.10 (s, 2H, OH), 8.82 (s, 2H, CHN), 5.58 (d, 2H, 2JHH 12.2, endo-CH2), 5.25 (d,
2H, 2JHH 14.1, endo-CH2), 4.84 (d, 2H, 2JHH 11.8, endo-CH2), 4.30 (overlapping d + t, 4H, ArCH2
+
endo-CH2), 3.95 (t, 2H, 2JHH 7.7 Hz, ArCH2), 3.73–3.27 (overlapping m, 12H, exo-CH2
+ NH2).
X-Ray crystallography for 2·2MeCN, 3·6MeCN, and 4·5MeCN
Intensity data were collected using Bruker SMART 1K CCD diffractometers. Sealed-tube Mo-Kα radiation (λ = 0.71073 Å) was used for 3·6MeCN, otherwise, synchrotron radiation (λ = 0.6923 Å for 2·2MeCN, λ = 0.6878 Å for 4·5MeCN) at Daresbury SRS Station 9.8, as described previously.7 Two-fold positional disorder was modelled with restraints for some calixarene p-tBu groups in 3·6MeCN and 4·5MeCN. Programs used: SHELXTL8 for structure solution and refinement and molecular graphics, Bruker AXS SMART (control), and SAINT (integration) and local programs.9Crystal data for 2·2MeCN: C66H72MoN4O4·2C2H3N, M = 1163.32, monoclinic, space group P21/n, a = 14.142(2), b = 21.946(3), c = 20.008(3) Å, β = 90.146(3)°, V = 6209.5(16) Å3, T = 160 K, Z = 4, μ = 0.264 mm−1, 29388 data measured, of which 10529 were unique, Rint = 0.189, all unique data used in refinement against F2 values to give final wR = 0.2341 (for all data), R = 0.0926 [for 5448 data with F2 > 4σ(F2)]. The structure suffered from ca. 58 : 42(2) twinning with the b and c axes interchanged. For 3·6MeCN: C116H132Mo2N4O8·6C2H3N, M = 2148.46, trigonal, space group P32, a = 16.7686(15), c = 34.515(3) Å, V = 8404.9(13) Å3, T = 160 K, Z = 3, μ (Mo-Kα) = 0.286 mm−1, 44602 data measured, of which 19554 were unique, Rint = 0.093, all unique data used in refinement against F2 values to give final wR = 0.1429 (on F2 for all data), R = 0.0718 [for 13334 data with F2 > 4σ(F2)], absolute structure parameter x = − 0.01(3). For 4·5MeCN: C146H172Mo2N4O10.5C2H3N, M = 2540.03, monoclinic, space group P21/c, a = 23.3956(17), b = 31.216(2), c = 21.2594(15) Å, β = 107.908(2)°, V = 14773.7(18) Å3, T = 160 K, Z = 4, μ = 0.228 mm−1, 83968 data measured of which 31474 were unique, Rint = 0.079, all unique data used in refinement against F2 values to give final wR = 0.1869 (on F2 for all data), R = 0.0670 [for 18014 data with F2 > 4σ(F2)].
CCDC reference numbers 173184–173186. See http://www.rsc.org/suppdata/nj/b1/b108088c/ for crystallographic data in CIF or other electronic format.
Acknowledgements
The Leverhulme Trust (Special Research Fellowship to C. R.) and The University of East Anglia are thanked for financial support. The EPSRC are thanked for provision of the Bruker AXS SMART diffractometers at Newcastle University and at Daresbury Laboratory, and for the award of beam time at Daresbury. We would like to thank Prof. W. Clegg for use of the X-ray facilities at Newcastle University.References
- (a) D. M. Roundhill, Prog. Inorg. Chem., 1995, 43, 533 Search PubMed; (b) C. Wieser, C. B. Dieleman and D. Matt, Coord. Chem. Rev., 1997, 165, 93 CrossRef CAS.
- (a) A. Zanotti-Gerosa, E. Solari, L. Giannini, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1998, 120, 437 CrossRef CAS; (b) L. Giannini, E. Solari, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1999, 38, 1438 CrossRef CAS; (c) L. Giannini, E. Solari, A. Zanotti-Gerosa, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Angew. Chem., Int. Ed. Engl., 1997, 36, 753 CrossRef CAS; (d) B. Castellano, E. Solari, C. Floriani, R. Scopelliti and N. Re, Inorg. Chem., 1999, 38, 3406 CrossRef CAS; (e) A. Zanotti-Gerosa, E. Solari, L. Giannini, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Chem. Commun., 1997, 183 RSC; (f) L. Giannini, E. Solari, A. Zanotti-Gerosa, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Angew. Chem., Int. Ed. Engl., 1996, 35, 85 CrossRef CAS; (g) B. Castellano, A. Zanotti-Gerosa, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1996, 15, 4894 CrossRef CAS; (h) L. Giannini, A. Casella, E. Solari, C. Floriani, A. Chiesi-Villa, C. Rizzoli, N. Re and A. Sgamellotti, J. Am. Chem. Soc., 1997, 119, 9198 CrossRef CAS.
- (a) M. Fan, H. Zhang and M. Lattman, Chem. Commun., 1998, 99 RSC; (b) F. J. Parlevliet, A. Olivier, W. G. J. de Lange, P. C. J. Kamer, H. Kooijman, A. L. Spek and P. W. N. M. van Leeuwen, Chem. Commun., 1996, 583 RSC; (c) S. G. Bott, A. W. Coleman and J. L. Atwood, Chem. Commun., 1986, 610 RSC; (d) G. E. Hofmeister, F. E. Hahn and S. F. Pedersen, J. Am. Chem. Soc., 1989, 111, 2318 CrossRef CAS; (e) G. E. Hofmeister, E. Alvarado, J. A. Leary, D. I. Yoon and S. F. Pedersen, J. Am. Chem. Soc., 1990, 112, 8843 CrossRef CAS; (f) V. C. Gibson, C. Redshaw, W. Clegg and M. R. J. Elsegood, Chem. Commun., 1995, 2371 RSC; (g) F. J. Parlevliet, M. A. Zuideveld, C. Kiener, H. Kooijman, A. L. Spek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1999, 18, 3394 CrossRef CAS; (h) C. Redshaw and M. R. J. Elsegood, Inorg. Chem., 2000, 39, 5164 CrossRef CAS; (i) J. M. Harrowfield, M. I. Ogden, W. R. Richmond and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 2153 RSC; (j) J. M. Harrowfield, M. I. Ogden and A. H. White, J. Chem. Soc., Dalton Trans., 1991, 2625 RSC; (k) P. Thuéry, M. Nierlich, M. I. Ogden and J. M. Harrowfield, Supramol. Chem., 1998, 9, 297 Search PubMed; (l) L. M. Engelhardt, B. M. Furphy, J. M. Harrowfield, D. L. Kepert, A. H. White and F. R. Wilner, Aust. J. Chem., 1988, 41, 1465 CAS; (m) J. M. Harrowfield, M. I. Ogden and A. H. White, Aust. J. Chem., 1991, 44, 1237 CAS; (n) J. M. Harrowfield, M. I. Ogden and A. H. White, Aust. J. Chem., 1991, 44, 1249 CAS; (o) B. M. Furphy, J. M. Harrowfield, D. L. Kepert, B. W. Skelton, A. H. White and F. R. Wilner, Inorg. Chem., 1987, 26, 4231 CrossRef CAS; (p) P. Thuery, N. Keller, M. Lance, J. D. Vigner and M. Nierlich, Acta Crystallogr., Sect. C, 1995, 51, 1570 CrossRef; (q) L. Le Clainche, M. Giorgi and O. Reinaud, Inorg. Chem., 2000, 39, 3436 CrossRef CAS; (r) C. D. Andreeti, G. Calestani and F. Ugozzoli, J. Inclusion Phenom., 1987, 5, 123 CrossRef; (s) Z. Asfari, A. Bilyk, J. W. C. Dunlop, A. K. Hall, J. M. Harrowfield, M. W. Hosseini, B. W. Skelton and A. H. White, Angew. Chem., Int. Ed., 2001, 40, 721 CrossRef CAS.
- V. C. Gibson, C. Redshaw, W. Clegg, M. R. J. Elsegood, U. Siemeling and T. Türk, J. Chem. Soc., Dalton Trans., 1996, 4513 RSC.
- V. C. Gibson, C. Redshaw, W. Clegg and M. R. J. Elsegood, Chem. Commun., 1998, 1969 RSC.
- K. J. C. van Bommel, W. Verboom, R. Hulst, H. Kooijman, A. L. Spek and D. N. Reinhoudt, Inorg. Chem., 2000, 39, 4099 CrossRef CAS.
- (a) W. Clegg, M. R. J. Elsegood, S. J. Teat, C. Redshaw and V. C. Gibson, J. Chem. Soc., Dalton Trans., 1998, 3037 RSC; (b) W. Clegg, J. Chem. Soc., Dalton Trans., 2000, 3223 RSC.
- G. M. Sheldrick, SHELXTL user manual, version 5, Bruker AXS Inc., Madison, WI, 1994 Search PubMed.
- SMART and SAINT software for CCD diffractometers, Bruker AXS Inc., Madison, WI, 1994.
Footnote |
| † First presented at the 221st ACS National Meeting, San Diego, 1–5th April 2001. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2002 |