Photonic antenna system for light harvesting, transport and trapping
Gion
Calzaferri
*a,
Marc
Pauchard
a,
Huub
Maas
a,
Stefan
Huber
a,
Abderrahim
Khatyr
a and
Tjeerd
Schaafsma
b
aDepartment of Chemistry and Biochemistry, University of Berne, Freiestr. 3, 3012, Berne, Switzerland. E-mail: gion.calzaferri@iac.unibe.ch
bLaboratory of Biophysics, Department of Agrotechnology and Food Sciences, Wageningen University, Dreijenlaan 3, 6703 HA, Wageningen, The Netherlands
First published on 20th September 2001
Abstract
Host–guest composites with photonic antenna properties are described. The material consists of cylindrical zeolite L crystals the channels of which are filled with chains of joined but electronically non-interacting dye molecules. Light shining on a crystal is first absorbed and the energy is then transported by the dye molecules inside the tubes to the desired part. Data on crystals in the size range of 30 nm up to 3000 nm are reported. The synthesis principle we are using is based on the fact that molecules can diffuse into individual channels. This means that given the appropriate conditions, they can also leave the zeolite by the same way. In some cases, however, it is desirable to block their way out, for stability reasons. This is done by adding a closure molecule. The general approach to connect the antenna function to its surroundings is to add “stopcock” molecules which generally consist of a head, a spacer and a label. They can either trap excitation energy on the external surface or inject excitation energy into the dye-loaded crystal. The stopcock molecules act as bridges between the dye molecules inside the channels and the outside world. Functionalisation of the closure and the stopcock molecules is an option for tuning e.g. wettability, refractive index, and chemical reactivity.
The wide-ranging tunability of the dye–zeolite L composites makes them useful for many applications. We discuss demonstration experiments which show the process of energy transfer and energy migration as educational tools, applications as high quality and non-toxic pigments, use as strongly luminescent pigments applicable as colour-changing media for LEDs, options for realising nanoscaled laser materials, and finally the challenge for realising solid state solar cells based on sensitisation of a thin semiconductor layer by energy transfer, the reversal of which can also lead to a new generation of LEDs.
1. Introduction
A photonic antenna is an organised multi-component arrangement in which several chromophores absorb the incident light and channel the excitation energy to a common acceptor component. Imaginative attempts to build an artificial antenna different from ours have been presented in the literature. Multinuclear luminescent metal complexes, multichromophore cyclodextrins, Langmuir–Blodgett films, dyes in polymer matrices and dendrimers have been investigated. Sensitisation processes in silver halide photographic materials and also the spectral sensitisation of polycrystalline titanium dioxide films in some cases show aspects of artificial antenna systems (for references see ref. 1). The materials which have been reported by us so far are of a bi-directional type. They are based on zeolite L as a host, and able to collect and transport excitation energy. 1–5 The transport is enabled by specifically organised dye molecules which mimic the natural function of chlorophyll. The zeolite L crystals consist of a continuous one-dimensional tube system. We have filled each tube with chains of joined but electronically non-interacting dye molecules. Light shining on the cylinder is first absorbed and the energy is then transported by the dye molecules inside the tubes to a desired region.The principle of two types of photonic antenna materials is illustrated in Fig. 1. The rectangles represent sites for dye molecules. Light gray rectangles are sites which contain donors while the dark rectangles contain acceptors. Both donors and acceptors are assumed to be strongly luminescent dyes. The dye molecule in Fig. 1a) which has been excited by absorbing an incident photon, transfers its electronic excitation to a neighbouring dye molecule. After a series of energy migration steps the electronic excitation reaches an acceptor. Energy migration is in competition with spontaneous emission, radiationless decay, and photochemically induced degradation. Very fast energy migration is therefore crucial if an acceptor should be reached before other processes take place. The energy quantum can be guided to a reaction centre once it has been captured by the acceptor. These conditions impose not only spectroscopic but also decisive geometrical constraints on the system. Recently we have been able to reverse the scheme in Fig. 1a) having acceptor dyes in the centre, and the donors at both ends, as illustrated in Fig. 1b). The material was investigated by stationary energy migration experiments on an ensemble and space and time resolved measurements on single crystals. 6
 | ||
| Fig. 1 Representation of a cylindrical nanocrystal consisting of organised dye molecules acting as donors (light grey rectangles) and acceptors (dark grey rectangles). a) The donors are in the middle part of the crystal and the acceptors at the front and the back of each channel. b) The donors are at the front and the back of each channel and the acceptors are in the middle part. The enlargements show details of a channel with a dye and its electronic transition moment (arrow) which is parallel with respect to the channel axis for long molecules and bent for shorter ones. The diameter of the channel windows is 0.71 nm and the largest free diameter is 1.26 nm. The centre to centre distance between two channels is 1.84 nm. | ||
The general synthesis concept of this inorganic–organic composites is based on the specific geometrical constraints imposed by the parallel arrangement of one-dimensional channels of the host. 4,5 Various synthesis strategies for the preparation of chromophores in porous silica and minerals have been reviewed recently. 7 Zeolite L crystals usually have cylindrical morphology, as illustrated in Fig. 2, where we also show a schematic view of their channel structure. The number of parallel channels which coincide with the c-axis of the hexagonal framework is equal to 1.07 rcyl2, where rcyl is the radius of the crystal in nm. The length of a unit cell along the c-axis is 0.75 nm. This means that a crystal of e.g. 600 nm diameter and 300 nm length gives rise to about 100000 parallel channels, each consisting of 400 unit cells (u.c.). 8–11
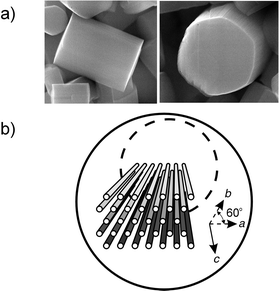 | ||
| Fig. 2 a) Side and top views of zeolite L crystals with a length of 950 nm and a diameter of 850 nm, respectively. b) A schematic view of some channels in a hexagonal zeolite crystal with cylindrical morphology. This is a pre-publication version of figures to be included in a forthcoming work tentatively titled Advances in Photochemistry Volume 27 edited by D.C. Neckers, to be published in 2002. Copyright © 2001 John Wiley & Sons Inc. All rights reserved. | ||
Control of the shape and size of the crystals is a necessary prerequisite for particular applications. Large crystals of a few hundred to a few thousand nm are very useful for studying the optical and photophysical properties of dye–zeolite composites on single crystals by means of optical microscopy methods. Crystals in the range of a few tenths to a few hundred nm are needed for high efficiency photonic antenna materials, useful as fluorescent microprobes in cell biology and analytical chemistry, 12 for developing a new generation of dye sensitised solid state solar cells, 13 or for preparing a new generation of light emitting diodes. We have recently shown how fine tuning of the size of zeolite L crystals in the size range of 30 nm up to 3000 nm can be realised by changing the composition of the starting gel for otherwise constant reaction conditions. It was thus possible to extend the investigations on energy migration in Py +-loaded zeolite L crystals, modified with Ox + as a luminescent acceptor at the ends of the crystals; see Table 1 for the structuresd of these molecules. 1,5
| a From ref. 14. | |||
|---|---|---|---|
| BP |

|

|
Py + |
| pTP |

|

|
PyGY + |
| DPH |

|

|
PyB + |
| PBOX |

|

|
Ox + |
| MBOXE |

|

|
Th + |
| POPOP |

|

|
ResH |
| DMPOPOP |

|

|
Hydroxy-TEMPO |
| DSC |

|
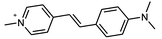
|
DMSI + |
| Fluorenone |
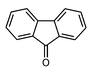
|

|
N-Ethylcarbazole |
| Naphthalene a |

|

|
Anthracene a |
Some dye molecules which have been inserted into zeolite L are given in Table 1 where we also give abbreviations used in this article. It is important to distinguish between three types of dye molecules. (i) Molecules small enough to fit into a single unit cell. Some examples are biphenyl (BP), hydoxy-TEMPO, fluorenone, and naphthalene. (ii) Molecules the size of which makes it hard to guess if they align along the c -axis or if they find a way to fit into a single unit cell. Ox +, Py +, and Th + are molecules of this type. 15 (iii) Molecules which are so large that they have no other choice but to align along the c -axis. Many examples fit into this category. POPOP, DMPOPOP, MBOXE, pTP, and DPH are among these. It is important to know more precisely which of the type (ii) and (iii) molecules can arrange in a way so that they can interact via their π-systems and which are only in physical contact, i.e. have negligible electronic interaction.
2. Energy migration
The two cationic dyes Py +, as a donor, and Ox +, as an acceptor, were found to be very versatile to demonstrate photonic antenna functionalities for light harvesting, transport, and capturing, as illustrated in Fig. 3. They can be incorporated into zeolite L by means of ion exchange, where they are present as monomers because of the restricted space. In this form they have a high fluorescence quantum yield and favourable spectral properties. The insertion of the dyes can be visualised by means of fluorescence microscopy. The fluorescence anisotropy of Ox +-loaded zeolite L has recently been investigated in detail by conventional and by confocal microscopy techniques. 15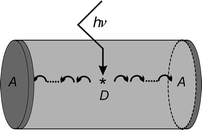 | ||
| Fig. 3 Simplified view of a bi-directional photonic antenna. The middle part shown in light grey contains donor molecules D. After excitation of D to D* the excitation energy migrates with equal probability to the left and to the right until it reaches an acceptor A (dark grey) which captures the excitation energy and emits it as red-shifted light. | ||
The occupational probability p of the sites with a dye is equal to the number of occupied sites divided by the number of sites available. Using this notion the Förster type energy transfer 16,17 and the energy migration rate constant k ij from a molecule i to a molecule j in this material can be expressed by: 2
 | (1) |
We describe properties of Py +-loaded zeolite L of which the channel ends are modified with Ox +. Ox + acts as an acceptor which becomes excited via radiationless energy transfer from an excited Py + moiety. If radiationless relaxation is not considered, Ox + can lose its excitation energy only by luminescence, as it cannot transfer the energy back to Py + because of its lower value. Fluorescence of excited Py +, internal conversion, and intersystem crossing compete with the energy migration and energy transfer processes. The efficiency of energy migration among the Py + molecules along the crystal can be investigated by measuring the front–back trapping efficiency TFB,∞. The front–back trapping efficiency is equal to the ratio of fluorescence intensity emitted by the acceptor Ox + ( IA) divided by the total fluorescence intensity ( IA + ID). 2,18
 | (2) |
We show in Fig. 4 experimental results obtained with crystals of the following average lengths: A, 300 nm; B, 500 nm; C, 850 nm; D, 1400 nm; and E, 2400 nm. These crystals were loaded with Py + in such a way that the occupational probability was always the same, namely ppy = 0.11. They were then modified with two Ox + molecules on average at both ends of each channel. The fluorescence of a thin layer on quartz was measured at room temperature after specific excitation of Py + at 460 nm. It shows a strong increase of the Ox + emission with decreasing crystal length lcyl. The front–back trapping efficiency increases from 0.33 up to 0.91. This means that in the 300 nm crystals, 91% of the emitted light is due to energy migration among the Py + molecules and finally transfer to the luminescent acceptor Ox +. Experiments with crystals in the size range of 50 nm show a similar behaviour.
 | ||
| Fig. 4 a) EM pictures of the investigated zeolite L samples with different mean crystal lengths lcyl: A, 300 nm; B, 500 nm; C, 850 nm; D, 1400 nm; E, 2400 nm (the bars in A–D correspond to 1 μm, in E to 3 μm). b) Fluorescence intensity (scaled to the same height at the maximum of the Py + emission) of Py +-loaded and Ox + modified zeolite L crystals with constant Py + loading ( ppy = 0.11) as a function of crystal length after specific excitation of only Py + at 460 nm. The Ox + modification was, on average, two molecules at both ends of each channel. 5 | ||
A small shift of the Py + maximum, from 525 nm for the smallest crystals to 530 nm for the largest ones, is observed. This wavelength shift may be due to self-absorption and re-emission because the absorption depth increases with increasing crystal size despite the constant ppy. The maximum of the Ox + emission remains at 605 nm.
The time evolution of the excitation distribution along the cylinder axis of this material is of great interest. 1,2,6 We report theoretical results obtained for a crystal consisting of 90 slabs, each of which corresponds to the thickness of a site. The photophysical data of Py + as donor and Ox + as acceptor were used to calculate the excitation distribution of the donors. Immediately after excitation all slabs have the same excitation probability. The excitation distribution is shown after 5, 10, 50 and 100 ps. We observe in Fig. 5a) that the slabs close to the acceptor layers lose their excitation energy very quickly. The trapping rate is proportional to the gradient of the excitation distribution at the position of the acceptors. Hence, it depends not only on the remaining excitation probability but also on the excitation distribution. This is in contrast to the donor fluorescence rate, which depends only on the excitation distribution of the donors. The fluorescence rate of the acceptors is proportional to their excitation probability. We therefore expect a fluorescence decay behaviour as illustrated in Fig. 5b). The fluorescence decay of the donors becomes much faster in the presence of acceptors, because of the depopulation due to the irreversible energy transfer to the acceptors. The fluorescence intensity stemming from the acceptors initially increases because excited states must first be populated via energy transfer from the donors. It therefore reaches a maximum before it decays.
 | ||
| Fig. 5 a) Excitation distribution along the channel axis of a zeolite L crystal consisting of 90 slabs (occupational probability p = 0.3) under the condition of equal excitation probability at t = 0 calculated for front–back trapping. Fluorescence of the donors is taken into account. A: t = 5 ps, B: t = 10 ps, C: t = 50 ps, and D: t = 100 ps after irradiation. b) Fluorescence decay of the donors in the absence of acceptors (dotted), in the presence of acceptors at both ends (solid), and fluorescence decay of the acceptors (dashed). | ||
A sophisticated bi-directional antenna material with three dyes is illustrated in Fig. 6. 4 After selective excitation of dye1, located in the middle part, the light energy is carried spectrally from the blue to green (dye2) to red (dye3) and spatially from the crystal centre to its left and right ends. The stacking of the dyes in the crystal can be seen in microscopy images taken on relatively large crystals ( Fig. 6b)). We show this for a 2000 nm crystal with POPOP in the middle, followed by Py + and then by Ox +. The different colour regions which can be observed in this experiment are impressive. Image A shows the fluorescence observed after selective excitation of the POPOP. The middle of the crystal shows the blue fluorescence of POPOP while the red emission of Ox + appears at both ends. Between these two zones the superposition of the fluorescence of all three dyes results in emission of white light. In image B the fluorescence is shown after selective excitation of the Py +. The red emission, hardly visible at both ends, and the green emission stem from the Ox + and the Py +, respectively. The yellow emission is due to superposition of the green and red fluorescence. Image C shows the emission of Ox + after selective excitation of only Ox +. These three microscopy images show very nicely the sequence of the inserted dyes in the channels, although the limited resolution does not allow one to observe completely separated regions. The microscopy pictures show how the resolution of the microscope decreases with longer wavelength of the observed light. Although the concentration of Ox + in the crystal is much lower than that of POPOP with the Ox + molecules located only at the very ends of the channels, the observed fluorescence in image C appears over a large area of the crystal.
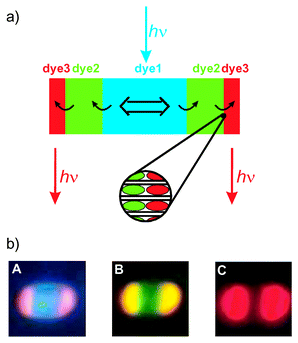 | ||
| Fig. 6 a) Principle of a bi-directional three-dye photonic antenna. The enlarged section shows the organisation of individual dye molecules at the domain boundary between dye2 and dye3. b) Fluorescence microscopy images of an Ox +,Py +,POPOP–zeolite L crystal of 2000 nm length upon selective excitation of A POPOP at 330–385 nm, B Py + at 470–490 nm, and C Ox + at 545–580 nm. The excitation light was eliminated by appropriate cut-off filters in front of the CCD-camera: A 410 nm, B 515 nm, C 605 nm. | ||
After this overview of photonic antenna properties we now turn to some intriguing optical properties we have recently observed in different dye–zeolite guest–host composites.
3. Optical properties
We describe observations made on dye-loaded zeolite L crystals of dimensions which are at least equal to or a few times larger than the wavelength of visible light. The refractive index of zeolites is in the same range as that of quartz. It is expected to vary to some extent depending on the cations, the water content, and the dye loading. Thus optical effects observed in tiny glass fibres are expected to appear in zeolite L crystals as well. The luminescent dyes inserted into the channels cause some new phenomena, also because of the pronounced anisotropy of their light absorption and emission properties and because the refractive index changes in regions of strong absorption. The orientation of the electronic transition moments of Ox + in zeolite L is 72° with respect to the c-axis while that of POPOP, DMPOPOP and similar molecules is parallel to this axis. 1,153.1 Fata morgana effects in dye-loaded zeolite L crystals
Optical effects due to refraction and total internal reflection have been observed in dye-loaded zeolite L crystals of 2.5 μm length and 1.4 μm diameter by means of an optical microscope equipped with polarisers, narrow band and cutoff filters. An astonishing effect taking place in an Ox +-loaded crystal is seen in Fig. 7. Looking at the polarised red emission, a homogeneous intensity distribution is observed over the whole crystal, with the exception of two perpendicular dark lines which separate the luminescence at both ends. The fluorescence in the middle part of the crystal decreases, when turning the polariser by 90°, but the two wings at both sides appear with about the same intensity as before. Obviously the light observed at both ends of the crystal is not polarised while that observed in the middle part is strongly polarised. Ox + molecules located at the outer surface of the crystals can be quantitatively destroyed by treating the material with a hypochlorite solution. 3,5 Such a treatment does not alter the optical properties of the material. The effect (dark lines and wings) disappears, however, upon refractive index matching, e.g. when applying an immersion oil. This means that the optical phenomenon is not due to some molecules present at the outer surface. We have to seek for another explanation. | ||
| Fig. 7 Polarised fluorescence microscopic pictures of a 2.5 μm long Ox +-loaded zeolite L crystal after excitation at 545–580 nm (cut-off 605 nm). The arrows indicate the transmission direction of the polariser. | ||
The luminous non-polarised wings are due to part of the emission built up in the middle of the crystal. They can be understood when taking total internal reflection into account. Refraction of the emission from the object O at the zeolite L/air interface observed at an angle α2 is explained in Fig. 8a). The emission can appear between 0° and the critical angle for total reflection α1,max, which is 42° for a refractive index of 1.49. The used objective of the microscope collects all light that is emitted under an angle α2 < 64° and therefore the object appears at the positions O′ in a circle with radius Δ l around the object O.
 | ||
| Fig. 8 Refraction and pathway of the emission from the object O hitting the zeolite L/air interface. a) Refraction of the emission from the object O at the zeolite L/air interface observed at an angle α2. The emission appears in the range between 0° and the critical angle for total reflection α1,max which is 42° ( d is the distance between O and the interface). b) Pathway of the emission of a molecule O. | ||
An incident photon hitting the wall at an angle α1 larger than 42° is totally reflected and can only leave the cylinder at its ends (see Fig. 8b)). In the case shown on the right side of Fig. 8b), it appears as luminous non-polarised wings. Hence, the emission observed in an optical microscope appears not at the origin of the molecule but outside of the crystal at both ends. This means that “we see” the emitting molecules at another place in space than they are, similarly to a mirage, which shows an image which may be hundreds of kilometres away, but can appear to be closer than it is. This phenomenon is called fata morgana. We are not sure if we understand the second part of the observation, namely the two dark regions which separate the polarised emission of the bulk from the non-polarised wings, but these may be due to interference phenomena.
Another nice fata morgana can be observed in DMPOPOP–zeolite L microcrystals, the luminescence of which is strongly polarised along the c-axis. Looking at standing crystals, a non-polarised blue ring with a dark centre is observed, see Fig. 9a). The dark part disappears immediately, the whole luminescent spot shrinks, and the luminescence intensity decreases when adding a drop of a solvent with the same refractive index as the zeolite. This shows that the blue ring is caused by refraction at the crystal/air interface.
 | ||
| Fig. 9 Fluorescence microscopic pictures of a standing 2.5 μm long DMPOPOP-loaded zeolite L crystal upon excitation at 330–385 nm and observed with a cut-off filter (410 nm), a) in air and b) in a refractive index matching solvent. | ||
Two kinds of refraction are responsible for the appearance of the luminescent ring with a dark spot in the middle. Emission at O which hits the wall at an angle between 17 and 42°, as shown in Fig. 10a), is refracted but can still be detected by the microscope. It appears to originate further away from the centre than it is in reality. As a consequence, the centre appears darker. Total reflection, as shown in Fig. 10b), causes a photon to travel on a helical pathway until it reaches the top or the bottom of the cylinder. If the photon encounters another excited molecule on its way, stimulated emission can occur. The information from where the emission originates is nearly totally lost. An angle of total reflection can only be realised by excited molecules which are near the side walls. This again makes the centre appear darker. The phenomenon seems to reveal the characteristics of a very tiny ring resonator which, however, perhaps resembles the ring resonator reported recently for much larger crystals. 19
 | ||
| Fig. 10 Refraction and pathway of the emission from the object O hitting the zeolite L/air interface. a) Emission at O which hits the wall at an angle between 17 and 42° can be detected by the microscope. Its refraction makes it appear to originate from the region O′, i.e. closer to the wall. b) Emission at O which hits the wall at an angle greater than 42° is totally reflected and can travel around the crystal until it reaches one of the two ends. | ||
3.2 Polarisation of the fluorescence
Dye molecules of interest for energy transport in our system, have an oblong form and a strong π–π*-transition with a transition moment parallel to the molecules’ long axis. The absorption and emission of light from these molecules is therefore strongly polarised. In an ensemble of many molecules this polarisation can only be observed when the molecules are ordered. Their width allows them to penetrate the one-dimensional channels of zeolite L. The geometrical constraints lead to an anisotropic organisation of the dyes and result in a net polarisation anisotropy.How exactly the molecules are oriented inside the channels depends on their specific shape and on the adsorption interaction between the dyes and the channel walls or charge compensating cations. Because of the dye’s oblongness a double-cone like distribution in the channels is a reasonable model. This distribution is illustrated in Fig. 11a). The arrows represent the transition moments of the dyes and α describes the half opening angle of the double cone. The hexagonal structure of the zeolite L crystal hence allows six equivalent positions of the transition moments on this double cone with respect to the channel axis.
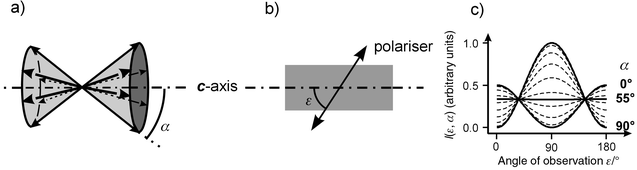 | ||
| Fig. 11 a) Distribution of the transition moments on a double cone with a half opening angle α. b) Polarisation direction observed when a single crystal is examined by means of a polariser. c) Relative intensity of the observed fluorescence as a function of the observation angle ε with respect to the crystal c-axis, for different half cone angles α. | ||
Fig. 11b) shows how the fluorescence polarisation of a single zeolite L crystal can be analysed by means of a polariser, which only transmits the indicated polarisation direction. In Fig. 11c) the theoretical relative intensity of the observed fluorescence is plotted versus the angle ε between the observed polarisation and the crystal c-axis for different values of α. If α is equal to 0°, the molecules’ transition moment coincides with the crystal c-axis and maximum fluorescence can be observed at ε equal to 0 or 180°. Such a crystal emits most of its fluorescence parallel to the crystal axis and remains dark if the polariser is set perpendicular to it. If α is equal to 90°, then the maximum fluorescence is observed perpendicular to the c-axis and the crystal emits no light parallel to it. For all α values between 0 and 90° there is a gradual change in behaviour leads from one extreme to the other. The differences between the maximum and minimum fluorescence intensity is reduced and at a magic angle of 54.7° no fluorescence anisotropy can be observed although the transition moments are not randomly oriented in the crystal.
In the case of Ox + in zeolite L a half cone angle α of 72° was obtained from quantitative measurements on single crystals. 15 The orientation of the transition moments with respect to the zeolite channels can be determined directly from the fluorescence polarisation whereas the orientation of the transition moments with respect to the molecular axis can not be determined from these experiments. Strong electric fields can change the orientation of the dipole moments; i.e. the Stark effect. 20 Geometrical estimates of the maximum angle of the double cone in the case of Ox + led to the conclusion that the angle α for the molecules can not be larger than 40°. The observation of α = 72° for the transition moments was interpreted by the existence of a remarkable Stark effect in these materials. 15 The arguments do not apply for molecules aligned along the channel axis.
In Fig. 12 fluorescence microscopy images of different dye-loaded zeolite L single crystals are shown. Each line consists of three images of the same sample, but with different polarisation of the fluorescence observed. In the first one the total fluorescence of the crystals is shown and in the others the fluorescence with the polarisation direction indicated by the arrows is displayed. The zeolite was loaded with the following dyes: A Py +, B PyGY +, C PyB +, D POPOP (see Table 1). Most crystals show a typical sandwich structure with fluorescent dyes at the crystal ends and a dark zone in the middle. This situation can be observed when the diffusion of the dyes in the channels has not yet reached its equilibrium situation. It illustrates nicely how the molecules penetrate the crystals via the two openings on each side of the one-dimensional channels.
 | ||
| Fig. 12 Fluorescence microscopy images of dye-loaded zeolite L single crystals, without polariser (first column) and with polariser (second and third column: the direction of the transmitted polarisation is indicated by the arrows). Samples: A Py +–zeolite L, B PyGY +–zeolite L, C PyB +–zeolite L, D POPOP–zeolite L. | ||
Py + is a molecule with similar dimensions as Ox + and therefore the behaviour of the fluorescence polarisation is the same as already described for the Ox +–zeolite L sample: maximum fluorescence perpendicular to the crystal axis; minimum fluorescence parallel to it. PyGY + is slightly larger than Py +, because of the methyl groups. The observed fluorescence polarisation in this sample changes significantly compared to Py +. The maximum fluorescence intensity is now measured parallel to the c-axis and the difference between maximum and minimum intensity is rather small. According to Fig. 11c) this is an indication that the transition moments have changed to an angle α below 55°. In the case of PyB + (with four ethyl groups) the intensity difference between the minimum and maximum intensity is even more pronounced indicating an angle α near 0°. In the final images of this series the fluorescence of POPOP–zeolite L crystals is analysed. POPOP has an estimated molecular length of 19 Å and therefore occupies approximately 3 u.c. The fluorescence polarisation in the images D shows very nicely that the transition moments of the POPOP molecules are arranged parallel to the c-axis, which means α is equal to 0°.
4. Further developments
In natural photosynthesis, complex arrays of antennae collect the solar energy and convert it into chemical potential energy that drives the chemistry of the photosynthetic machinery. The processes involved are very fast and highly efficient. 21–23The dye–zeolite composites reported so far show fascinating photonic antennae properties which are perhaps comparable to some extent to those of natural systems. Tuning their chemical and photochemical behaviour, organising information exchange between their inside and the external world, but also organising individual crystals on a surface in order to realise e.g. mono-directional functionalities remains a challenge which we address in this section.
4.1 Closure and stopcock molecules
The synthesis principle we are using is based on the fact that molecules can diffuse into individual channels. 3,4,24 This means that under appropriate conditions, they can also leave the zeolite in the same manner. In some cases, however, it is desirable to block their way out, so as to stabilise the structure. This can be done by adding a 'closure' molecule after the synthesis has been completed. A variety of closure molecules which seal the channels completely or only partially can be used, depending on the requirement. An example already used is the addition of fluorenone which enters readily but leaves the structure reluctantly. If more complete sealing is needed molecules bearing appropriate reactive groups can be used. Functionalisation of the closure molecules is an option for tuning wettability, refractive index, chemical reactivity, and other properties.External trapping and injection of quanta is more demanding. The general approach we are using to solve this problem is to add “stopcock” molecules as illustrated in Fig. 13. 1 A stopcock molecule generally consists of three components: a head, a spacer and a label. The tail moiety (spacer + label) has a longitudinal extension of more than one u.c. along the c-axis. The head moiety has a lateral extension that is larger than the channel width and prevents the head from penetrating into the channel. The channels are therefore terminated in a generally plug like manner.
 | ||
| Fig. 13 a) Typical shape of a stopcock molecule located at the end of a zeolite channel. b) Fluorescent molecules which have already been inserted in zeolite L are modified with an inert head in order to build stopcock molecules with fluorescent tails. c) Examples of molecules which can be used as stopcocks with a fluorescent head. | ||
The main function of the stopcocks is to connect the antenna function of the crystal to its surroundings. They act as bridges between the dye molecules inside the channels and the outside world by either trapping excitation energy coming from the inside, or injecting excitation energy into the dye-loaded zeolite crystal. Stopcock-traps therefore have a sufficiently large spectral overlap integral with donor molecules inside the channels, while stopcock-donors have a large spectral overlap integral with acceptor molecules inside the channels.
Strongly fluorescent molecules are suitable for these energy transport functions. Since fluorescence is usually quenched by dimerisation 25,26 the design must be such that the chromophores do not electronically interact with each other. Both heads and labels can be fluorescent, depending on the needs. Fluorescent labels have the advantage of being protected by the zeolite framework. They are stabilised chemically and cannot form quenching dimers due to lack of space. Fluorescent heads on the other hand are closer to the external surface which is desirable for coupling the antenna to a device.
The labels are based on organic and silicon–organic backbones. The simplest form of a label is non-reactive. Its only function is to enter and to hook to the channel. Non-reactive labels can be either neutral or cationic. Reactive labels enter the zeolite channels and then undergo a chemical reaction under the influence of irradiation, heat or a sufficiently small reactant. In this way they hook themselves inside the zeolite channels or maybe bind to molecules already present.
The heads of the stopcock molecules are too large to be able to enter the channels. Typical functionalised groups such as bucky balls, 27 chelating centres 28 and others can be used. Some examples are given in Fig. 14. Smilarly to the labels, heads can be reactive or non-reactive. Reactive heads may have “arms” which can interact with each other to form a “monolayer polymer” or bind to the zeolite external surface.
 | ||
| Fig. 14 Schematic representation of several head moieties: a) fluorescent chemosensor, 75 b) C 60-based heads, 27 c) ligands containing suitable binding sites in a correct arrangement for metal ion complexation, 28 d) head moieties functionalised with reactive sites. 32 | ||
In some cases it is desirable to add a spacer which elongates the stopcock molecule so that the length of the tail can be controlled. This can be a tool to improve the solubility of the whole molecule. Polar groups might help to bind the molecules more strongly inside the zeolite channels. Spacers which are sufficiently flexible so that they can bend the tail into the zeolite channels include e.g. aliphatic chains, polyethers or amides.
Positively charged stopcocks can be plugged in the zeolite channels by ion exchange while neutral stopcocks can be added by dehydration of the zeolite channels and adsorption from a non-aqueous solution or from the gas phase. The zeolite’s external surface consists of a cylinder coat and two edges. These two surfaces differ in a number of properties so that the interactions can be tuned. For MFI type zeolites, as an example, it was reported that guest molecules bind to the holes on the external surface much more strongly than on the framework between the holes. 29
The specific coupling of the photonic antenna via the stopcocks to a target depends on the functionality envisaged. Direct linkage to a semiconductor surface, embedding into a semiconducting polymer, organisation of crystals bearing ionic stopcocks by means of charged polymers, 30,31 or linkage to sites of biological interest are possibilities.
4.2 Mono-directional antenna materials
In mono-directional antenna systems the energy is transported in one direction only. We discuss the possibilities sketched in Fig. 15 to create asymmetrical antenna materials. Either the insertion of the dyes into the zeolite channels is controlled such that the molecules can penetrate the channels only from one side, or crystals containing different dyes are organised appropriately. | ||
| Fig. 15 Schematic view of three strategies for the preparation of mono-directional antenna materials. a) Organisation of zeolite L crystals on a substrate. The channels are oriented perpendicular to the surface. Dye molecules can only be inserted from one side. b) Multilayer organisation of charged polymers and zeolite L crystals loaded with different dyes. c) Multilayer organisation of oppositely charged zeolite L crystals. | ||
Asymmetric loading of zeolite L crystals is possible if the channel openings are selectively closed on one side so that the dyes can be inserted from the other side in sequence by the same procedure as used for the preparation of the bi-directional antenna materials. 4 Organising the crystals on a substrate, maybe by linking them covalently, 32 in order to inhibit the penetration of dye molecules from the substrate side, is a way to realise this. Microstructured substrates have been used to organise relatively large zeolite ZSM-5 crystals in a plane. 33 Other forces which have been used for this purpose are adhesion 34 and electric fields. 35 The morphology and the size of the crystals are important in all cases. ZSM-5 nanoscale seed crystals have been organised with their largest area surface on positively charged polymers on a gold surface. Calcination and growth of the silicalite then led to a densely packed and oriented polycrystalline layer. 36 Zeolite L crystals can be organised by modifying them with charged stopcock molecules. This leads to an anisotropic distribution of the zeolite surface charges and gives rise to attractive electrostatic interactions with an oppositely charged substrate. The crystal growth of AlPO-5 in the presence of floating anodised alumina can be controlled to obtain vertically aligned hexagonal crystals. 37
To further suppress the insertion of dyes into the channels from the substrate-side, the channels can be sealed on one side by adsorption of a polymer onto the substrate. 38 Alternatively, stopcocks can be functionalised specifically with electro-reactive constituents like ethynylic-, 39 vinylic- 40 or pyrrolic-groups. 41,42 The substrate serves as electrode under the influence of an electrochemical potential and only the adjacent heads of the stopcocks react to form a covalently cross linked structure. The stopcocks are then removed from the other side of the crystals to unblock the channels for the dye insertion.
The formation of mono-directional antenna materials by organising different dye loaded crystals is illustrated in Fig. 15b) and c). Assembly of layered organic–inorganic composites is currently a major research area. 43,44 An increasing number of articles concerning the assembly of zeolite crystals on substrates via ionic or covalent bonding, some by the use of appropriate organic additives, have been published. 45–49 This knowledge can be used for alternate adsorption of charged polymers and zeolite L crystals modified with stopcocks bearing a charged head as illustrated in Fig. 15b). Polymeric materials ranging from biopolymers such as proteins 50,51 to inorganic macromolecules and clays 52,53 have been assembled by means of related techniques. Luminescent polymers can be used as connectors to transport the energy from one crystal to another one. 54 A simpler concept consists of arranging differently charged crystals in the manner shown in Fig. 15c).
5. Applications
The tunability in size and properties of the materials described in the previous sections is so large that we expect them to find applications in different fields such as optoelectronics, pigments, molecular probes but also as educational tools. We focus on five applications amongst these. Some of them are already feasible, while others require further development.5.1 Educational tools
There are not many demonstration experiments which show the process of energy transfer and energy migration well. We found that the energy transfer between dye molecules inside the channels of zeolite L works so well that it can be used as a nice educational tool to show both energy transfer and energy migration. The reason for this is that the dye molecules can be brought close together without forming dimers. We can vary the mean distance between the molecules by varying the occupational probability of the dyes which is directly proportional to the concentration of the dyes in the zeolite channels. The volume of a zeolite crystal is given by: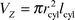 | (3) |
 | (4) |
 | (5) |
 | (6) |
 | (7) |
We concentrated on the two cationic dyes Py + and Ox + which can be incorporated into zeolite L from aqueous solution with about equal rates. Both dyes have a large fluorescence quantum efficiency and therefore radiationless relaxation is not considered. If a 1∶1 mixture of the two dyes is used we can look at the mechanism of energy transfer from Py + (donor D) to Ox + (acceptor A). We can also modify Py +-loaded zeolite L crystals with one molecule of Ox + on average at all channel endings. By varying the occupational probability of Py +, energy migration to the crystal endings can be tuned from poor to efficient.
The energy transfer in these materials can easily be observed when a series of samples is put in a black box as shown in Fig. 16. The samples were excited using a Mini Mag-Lite ® AA flashlight and a Schott DAD 8-1 interference filter at 486.7 ± 5 nm. The light beam has to be perpendicular to the interference filter and to the sample. The emission light is observed through a Schott OG 515 cut-off filter. The sample and the cut-off filter are not perpendicular to the observation angle so as to minimise reflection effects.
 | ||
| Fig. 16 Black box for observation of energy transfer and energy migration demonstration experiments. 1: Mag-Lite ® AA flashlight, 2: Schott DAD 8-1 interference filter at 486.7 ± 5 nm, 3: sample, 4: Schott OG 515 cut-off filter. | ||
The transmission spectra of the two filters and the absorption and the emission spectra of Py + and Ox + are plotted in Fig. 17.
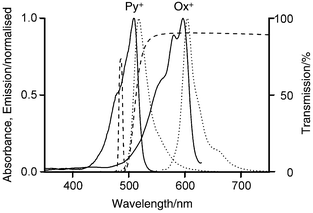 | ||
| Fig. 17 Absorption (solid) and emission spectra (dotted) of Py + and Ox + in zeolite L. The dashed lines show the transmission of the Schott DAD 8-1 interference filter and the Schott OG 515 cut-off filter. | ||
With a 1∶1 mixture of the two dyes we can observe the energy transfer from the Py + donors to the Ox + acceptors very clearly. The seven luminescent samples shown in Fig. 18 consist of zeolite L crystals of 300 nm average length containing different amounts of Py + and Ox +. In all cases Py + was specifically excited at 486.7 ± 5 nm.
 | ||
| Fig. 18 Photographic image of the fluorescence of dye-loaded zeolite L layers upon monochromatic irradiation at 485 ± 5 nm and observation through a 500 nm cut-off filter. 1 and 7 are references loaded with Py + and Ox + only (occupational probability 1.4 × 10 −2). 2– 6 contain a 1∶1 mixture of Py + and Ox + with the following occupational probabilities for each dye: 2, 6.8 × 10 −4; 3, 3.4 × 10 −3; 4, 6.8 × 10 −3; 5, 1.4 × 10 −2; 6, 2.8 × 10 −2. This is a pre-publication version of a figure to be included in a forthcoming work tentatively titled Advances in Photochemistry Volume 27 edited by D. C. Neckers, to be published in 2002. Copyright © 2001 John Wiley & Sons Inc. All rights reserved. | ||
Reference samples 1 and 7 contain only Py + or Ox +, respectively. The other samples are 1∶1 mixtures of the two dyes with increasing occupational probability. We can calculate the following mean donor–acceptor distances: 2, 187 Å; 3, 109 Å; 4, 87 Å; 5, 69 Å; 6, 55 Å. The Förster radius for Py + to Ox + energy transfer in a medium of refractive index of 1.4 is about 70 Å, based on the Py +/Ox + spectral overlap which is 1.5 × 10 −13 cm 3 M −1. In sample 2 we observe mainly the green luminescence of Py +, which means that the dyes are too far apart for Förster type energy transfer. The yellow colour of 3 is due to a mixture of green and red luminescence which means that energy transfer is significant. The energy transfer becomes more and more dominant, and by sample 6, the red luminescence of the Ox + is dominant.
One would expect that the colour of the samples 2– 6 in diffuse reflection mode to be the same since there is a 1∶1 mixture of the two dyes in all cases. This, however, is not observed because of the high concentration of dye molecules inside the zeolite. If the concentration of dye molecules inside the zeolite channels is high enough, a saturation effect is observed, i.e. light of specific wavelengths is totally absorbed and this changes the absorption spectrum as is shown in Fig. 19. Therefore the visual colour of the different samples observed (as diffuse reflection) changes from yellow to red with increasing loading.
 | ||
| Fig. 19 Calculated saturation effect for a 1∶1 mixture of Py + and Ox +. This effect changes the visual colour of the material. | ||
A similar experiment can be performed to show energy migration. One can modify Py +-loaded zeolite L crystals with, on average, one Ox + molecule at both ends of each channel. By varying the occupational probability of Py +, and thus the mean distance between the Py + molecules, the energy migration efficiency can be varied. If energy migration is efficient, the electronic excitation energy will be trapped at the crystal endings by the Ox + molecules and the red emission of Ox + molecules will be visible. If, however, the energy migration is not very efficient, the green emission of the Py + molecules will dominate.
5.2 Pigments
A promising field with potential for industrial application is the use of dye-loaded zeolites as pigments. 55 The encapsulation of dyes in zeolite L leads in general to a substantially increased stability of the organic molecules. The matrix protects them from oxidative agents and other reactive species and the spatial constraints can eliminate photoisomerisation reactions. Additionally the processing properties of such pigments only depend on the zeolite surface, which can be tuned to a large extent, and not on the specific dye characteristics.The use of highly fluorescent dyes leads to much brighter colours. In particular, the xanthene dyes (Ox +, Py +, PyGY +, PyB +) act as optical brighteners because the fluorescence in the visible region can be initiated by absorption in the UV-region of the spectrum. By the use of only two different dyes a large palette of different colours can already be generated (see Fig. 18). New interesting colour effects can be controlled via the energy transfer, saturation, and re-absorption phenomena in these materials. Applying the closure and stopcock molecules discussed in section 4.1 allows fine tuning of the specific properties of the pigments.
5.3 Colour-changing medium
A LED-display consists of a matrix of contacts made to the bottom and top surfaces of each light emitting element, or pixel. To generate a full-colour image, it is necessary to vary the relative intensities of three closely spaced, independently addressable pixels, each emitting one of the primary colours of red, yellow or blue. Several techniques have been proposed for producing the three colours in each pixel. One method is the use of a single blue or ultraviolet LED to pump organic fluorescent wavelength converters, also known as colour-changing media (CCM), as illustrated in Fig. 20. 56 | ||
| Fig. 20 Scheme of a pixel for generating full colour. The green and red colours are generated by transforming the emitted blue or UV-light from the LEDs with the help of colour-changing media. | ||
The dye–zeolite L antenna systems have ideal properties to act as colour-changing media in such an application. Due to the high concentration of dye monomers in the zeolite matrix, almost all incoming light can be absorbed within the dimensions of one monolayer of such crystals. 3 Through the high energy transfer efficiency reported in Fig. 4, up to 90% of the absorbed light can then be converted into light of a longer wavelength. The anisotropic arrangement of the dye molecules leads to spatially well defined emission of partially polarised light. The scattering of light at the crystal surface can be minimised by the use of nanocrystals or can be eliminated by embedding the material into a medium with the same refractive index.
5.4 Nanoscaled lasers
As we reported in section 3.1, refraction and total internal reflection can occur in dye-loaded zeolite L microcrystals. Therefore a bundle of light rays in e.g. a POPOP-loaded zeolite-composite can circulate inside the hexagon. If the emission can circulate often enough in the same volume and the loss of the zeolite L ring cavity is small enough, lasing should be possible.Laser activity was indeed recently reported in pyridine-2-loaded nanoporous AlPO-5 molecular sieve. 19 Like zeolite L, this compound consists of hexagonally arranged one-dimensional channels along the crystal long axis which can act as a matrix to arrange geometrically suitable dyes along the c-axis in a supramolecular manner. The thickness of the used AlPO-5 crystals ranged from 4.5 μm to 22 μm and the corresponding length from more than ten to a hundred μm. The authors assumed that the resonator is built up by total internal reflection leading to a ring cavity. However, using a resonator with a size of the wavelength or less, classical optics arguments might not be sufficient.
To overcome the lasing threshold, the light amplification has to be larger than unity. For this reason the loss from the cavity has to be small. While the length of one round-trip in the largest crystals we are using is 3 μm, it is about 13.5 μm for the smallest described AlPO-5 laser cavity. The consequences of this are not well understood yet.
To obtain the spectrum of modes in a first approximation one could fit an integral number of wavelengths into the beam pathway to get constructive interference after a round-trip. Using the same arguments as in ref. 19, the wavelength interval Δ λ can be calculated between two constructive modes of a POPOP-loaded zeolite L laser with a diameter of ≅1 μm giving a round-trip length of L ≅ 3 μm at an emission wavelength of 440 nm. From the relation below:
 | (8) |
In contrast to the AlPO-5 system we are able to fine tune the dye concentration in a broad range. Furthermore the morphology of the material does not change with different dye loadings, because the dyes are inserted into the previously synthesised zeolite L. For neutral molecules it is possible to vary the loading from p ≅ 0.001 molecules per site up to a nearly filled zeolite L with a loading of p ≅ 1, while for ionic dyes a maximal loading of p ≅ 0.4 molecules per site has been realised up to now. Therefore a very high optical density of monomeric aligned dye molecules can be reached, shortening the minimum optical pathway for laser action.
Depending on the size of an incorporated dye, the angle of the transition dipole moment to the c-axis lies between 0° for long molecules and 72° for smaller ones. Therefore if a small molecule is inserted into the channels of zeolite L, part of the emission will be parallel to the c-axis. Due to the flat and parallel ends of appropriately prepared zeolite crystals one can envisage to arrange crystals between two mirrors or to add a reflecting layer on individual crystals. This might lead to a microlaser with a plane parallel resonator. Apart from experimental difficulties the realisation of a dye-loaded zeolite L nanolaser appears to be feasible.
5.5 Dye sensitised solar cells and LEDs
The currently available solar cells are an attractive source of renewable energy. They are, however, still expensive for large scale applications. During the last decades some effort has been made to find a cheap and efficient alternative for crystalline silicon p/n junctions. Remarkable work has been done on developing p/n junctions with organic semiconductor materials. 57–59 More groups, however, have focussed on dye sensitisation of metal oxide semiconductors. 60–63 In this type of dye sensitised solar cells, the dyes have the function of absorbing light and subsequently injecting an electron in the metal oxide semiconductor. After electron injection, the dyes have to be regenerated, usually by means a redox couple. A different kind of dye sensitised solar cell was proposed by Dexter 64 in 1979. He described sensitisation of the semiconductor by energy transfer instead of electron injection, followed by the production of an electron–hole pair in the semiconductor ( Scheme 1). Although Dexter published this idea in 1979, only a few groups so far have tried to observe energy transfer from a dye to a semiconductor. 65–67 | ||
| Scheme 1 Citation of D. L. Dexter, J. Lumin., 1979, 18/ 19, 779. 64 b | ||
The dyes do not have to be regenerated in this case because they do not exchange electrons. Energy transfer to the semiconductor works well, if the distance between the donor and the semiconductor is in the order of the critical distance for Förster energy transfer. Only a very thin semiconductor layer is needed, because the electron–hole pairs form near the surface. The flexibility in tuning the energy of the donors is large and only limited by the energy gap of the semiconductor which must be equal to or smaller than the excitation energy of the donor. Electron transfer is prevented by introducing a insulating layer.
The antenna effect, as it is found in natural photosynthetic systems, is an attractive tool to increase light absorption of solar cells. Some of the work done on dye sensitisation of polycrystalline titanium dioxide shows aspects of antenna behaviour. 62,68–72 Most of the problems in the systems where an electron is injected into the semiconductor arise in the regeneration process of the dyes. If the principle of energy transfer is used instead of electron injection this regeneration process can be avoided. The photonic antenna material developed in our group is based upon energy migration and energy transfer and it is challenging to combine its properties with the ideas of Dexter to form an antenna-sensitised solar cell. It appears to be feasible to put small crystals with a relatively large diameter with their c-axis perpendicular to the surface of a semiconductor (see Fig. 21). In this case, of course, a one directional antenna as described in section 4.2 is needed. The excitation energy is transported to the edge of the crystal by energy migration. At the edge of the crystal energy transfer takes place from a stopcock molecule to the semiconductor over a certain distance. Tandem solar cells are more advanced high efficiency systems. 73 In this concept, one-directional antenna materials are placed between two n-type semiconductors with different band gaps, as shown in Fig. 22. Both n-type semiconductors generate electrons, but the band gap of the first is tuned to the red dye while the band gap of the second is tuned to the blue.
 | ||
| Fig. 21 Sensitised solar cell based on dye-loaded zeolite L antenna systems. The antenna systems absorb light and transport their energy mainly along the c-axis of the crystals to the semiconductor surface. Electron–hole pairs are formed in the semiconductor by energy transfer from the antenna system to the conduction band of the semiconductor. | ||
 | ||
| Fig. 22 Tandem solar cell where mono-directional antenna systems with blue, yellow and red dyes are placed between two n-type semiconductors with different band gaps. | ||
The system discussed so far can be reversed. If we reverse the current and put a voltage over the semiconductor, it is possible to transfer energy from the semiconductor to the acceptor-antenna, as shown in Fig. 23. This results in light emitting nanocrystals. The colour of the emitted light can be tuned by adapting the ratio of the blue, yellow and red emitting dyes. Conventional LEDs are excited electrically. In this case the excitons are statistically distributed over singlet and triplet states. According to literature, a quarter of the formed excitons are singlet excitons which are responsible for the electroluminescence, while three quarters are triplet excitons which mainly decay non-radiatively. 74 In the case of direct energy transfer from a semiconductor to dye-loaded nanocrystals, mostly the excited singlet states are filled, leading to higher luminescence efficiency.
 | ||
| Fig. 23 Light emitting nanocrystals. The electrical source supplies excitation energy that can be transferred from the semiconductor to the nanocrystals. The dyes inside the crystals lose their energy by fluorescence because of their high fluorescence quantum yield. | ||
6. Conclusions
We have shown that the basic concept of filling hexagonal crystals, which consist of one-dimensional channels with molecular diameter and tuneable length, with luminescent dyes leads to new inorganic–organic host–guest materials with fascinating properties and challenging options for chemical modifications. From this result applications in different fields of great practical interest may result. The concepts described are not limited to zeolite L as a host. A nice feature of this zeolite is, however, that neutral as well as cationic dyes can be inserted. For many dyes insufficient space is available for electronic overlap, so that they retain the properties of the monomers. Additionally, the entrance of the channels is chemically sufficiently different with respect to the surface, so that fine tuning of stopcock molecules is feasible. Only a few experiments have been conducted with the 30–100 nm material so far. This will soon change, however, because it offers challenging opportunities for sensor applications, as molecular probes, or for incorporation into photoconductive polymers.Acknowledgements
This work was supported by the Swiss National Science Foundation Project NFP47 (4047-057481) and by the Bundesamt für Energiewirtschaft Project 10441. We also thank René Bühler for his contribution to realise the experiments reported in section 5.1.References
- G. Calzaferri, H. Maas, M. Pauchard, M. Pfenniger, S. Megelski and A. Devaux, Adv. Photochem., 2001, 27, in press Search PubMed.
- N. Gfeller and G. Calzaferri, J. Phys. Chem. B, 1997, 101, 1396 CrossRef CAS.
- G. Calzaferri, D. Brühwiler, S. Megelski, M. Pfenniger, M. Pauchard, B. Hennessy, H. Maas, A. Devaux and U. Graf, Solid State Sci., 2000, 2, 421 CrossRef.
- M. Pauchard, A. Devaux and G. Calzaferri, Chem. Eur. J., 2000, 6, 3456 CrossRef CAS.
- S. Megelski and G. Calzaferri, Adv. Funct. Mater., 2001, 11, 277 CrossRef CAS.
- M. Pauchard, S. Huber, R. Méallet-Renault, H. Maas, R. Pansu and G. Calzaferri, Angew. Chem., Int. Ed., 2001, 40, 2839 CrossRef CAS.
- G. Schulz-Ekloff, D. Wöhrle, B. van Duffel and R. A. Schoonheydt, Microporous Mesoporous Mater., in press Search PubMed.
- D. W. Breck, in Zeolite Molecular Sieves, Wiley, New York, 1974 Search PubMed.
- W. M. Meier, D. H. Olsen and C. Bärlocher, in Atlas of Zeolite Structure Types, Elsevier, London, 1996 Search PubMed.
- T. Ohsuna, Y. Horikawa and K. Hiraga, Chem. Mater., 1998, 10, 688 CrossRef CAS.
- P. A. Anderson, A. R. Armstrong, A. Porch, P. P. Edwards and L. J. Woodall, J. Phys. Chem. B, 1997, 101, 9892 CrossRef CAS.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, ed. Kluver, Academic/Plenum, New York, 1999 Search PubMed.
- G. Calzaferri, Chimia, 1998, 52, 525 CAS.
- S. Hashimoto, M. Hagiri, N. Matsubara and S. Tobita, Langmuir, submitted Search PubMed.
- S. Megelski, A. Lieb, M. Pauchard, A. Drechsler, S. Glaus, C. Debus, A. J. Meixner and G. Calzaferri, J. Phys. Chem. B, 2001, 105, 25 CrossRef CAS.
- T. Förster, Ann. Phys. (Leipzig), 1948, 2, 55 Search PubMed.
- T. Förster, in Fluoreszenz Organischer Verbindungen, Vandenhoeck & Ruprecht, Göttingen, 1951 Search PubMed.
- N. Gfeller, S. Megelski and G. Calzaferri, J. Phys. Chem. B, 1999, 103, 1250 CrossRef CAS.
- I. Braun, G. Ihlein, F. Laeri, J. U. Nöckel, G. Schulz-Ekloff, F. Schüth, U. Vietze, Ö. Weiss and D. Wöhrle, Appl. Phys. B: Lasers Opt., 2000, 70, 335 CrossRef CAS.
- A. Renn, S. E. Bucher, A. J. Meixner, E. C. Meister and U. P. Wild, J. Lumin., 1988, 39, 181 CrossRef CAS.
- Z. G. Fetisova, A. M. Freiberg and K. E. Timpmann, Nature, 1988, 334, 633 CrossRef CAS.
- X. Hu and K. Schulten, Phys. Today, 1997, 28 CAS.
- G. D. Scholes and G. R. Fleming, J. Phys. Chem. B, 2000, 104, 1854 CrossRef CAS.
- M. Pfenniger and G. Calzaferri, ChemPhysChem, 2000, 4, 211 CrossRef.
- M. Kasha, H. R. Rawls and A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721 CrossRef CAS.
- D. M. Guldi, M. Maggini, E. Menna, G. Scorrano, P. Ceroni, M. Marcaccio, F. Paolucci and S. Roffia, Chem. Eur. J., 2001, 7, 1597 CrossRef CAS.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759 CrossRef CAS.
- N. J. Turro, X.-G. Lei, W. Li, Z. Liu, A. McDermott, M. F. Ottaviani and L. Abrams, J. Am. Chem. Soc., 2000, 122, 11649 CrossRef CAS.
- G. Ladam, P. Schaad, J. C. Voegel, P. Schaaf, G. Decher and F. Cuisinier, Langmuir, 2000, 16, 1249 CrossRef CAS.
- T. Cassier, K. Lowack and G. Decher, Supramol. Sci., 1998, 5, 309 CrossRef.
- K. Ha, Y.-J. Lee, H. J. Lee and K. B. Yoon, Adv. Mater., 2000, 12, 1114 CrossRef CAS.
- L. Scandella, G. Binder, J. Gobrecht and J. C. Jansen, Adv. Mater., 1996, 8, 137 CAS.
- P. Lainé, R. Seifert, R. Giovanoli and G. Calzaferri, New J. Chem., 1997, 21, 453 Search PubMed.
- J. Caro, G. Finger, J. Kornatowski, J. Richter-Mendau, L. Werner and B. Zibrowius, Adv. Mater., 1992, 4, 273 CrossRef CAS.
- S. Mintova, B. Schoeman, V. Valtchev, J. Sterte, S. Mo and T. Bein, Adv. Mater., 1997, 9, 585 CAS.
- T.-G. Tsai, K.-J. Chao, X.-J. Guo, S.-L. Sung, C.-N. Wu, Y.-L. Wang and H.-C. Shih, Adv. Mater., 1997, 9, 1154 CrossRef CAS.
- (a) J. Caro, B. Zibrowius, G. Finger, M. Buelow, J. Kornatowski and W. Huebner, Ger. Offen., DE 41 09 038 A1, 1992. Search PubMed; (b) J. Caro, B. Zibrowius, G. Finger and J. Richter-Mendau, Ger. Offen., DE 41 09 037 A1, 1992 Search PubMed.
- K. Shiga, T. Inoguchi, K. Mori, K. Kondo, K. Kamada, K. Tawa, K. Ohta, T. Maruo, E. Mochizuki and Y. Kai, Macromol. Chem. Phys., 2001, 202, 257 CrossRef CAS.
- J. M. Calvert, R. H. Schmehl, B. P. Sullivan, J. S. Facci, T. J. Meyer and R. W. Murray, Inorg. Chem., 1983, 22, 2151 CrossRef CAS.
- A. Deronzier and J. C. Moutet, Coord. Chem. Rev., 1996, 147, 339 CrossRef CAS.
- A. F. Diaz, K. K. Kanazawa and G. P. Gardini, J. Chem. Soc., Chem. Commun., 1979, 635 RSC.
- M. Zanetti, S. Lomakin and G. Camino, Macromol. Mater. Eng., 2000, 279, 1 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- Y. Yan and T. Bein, J. Am. Chem. Soc., 1995, 117, 9990 CrossRef CAS.
- Y. Yan and T. Bein, J. Phys. Chem., 1992, 96, 9387 CrossRef CAS.
- T. Bein, Chem. Mater., 1996, 8, 1636 CrossRef CAS.
- Z. Li, C. Lai and T. E. Mallouk, Inorg. Chem., 1989, 28, 178 CrossRef CAS.
- L. C. Boudreau and M. Tsapatsis, Chem. Mater., 1997, 9, 1705 CrossRef.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117 CrossRef CAS.
- W. Kong, X. Zang, M. L. Gao, H. Zhou, W. Li and J. C. Shen, J. Macromol. Rapid Commun., 1994, 15, 405 Search PubMed.
- N. A. Kotov, I. Dékány and J. H. Fendler, J. Phys. Chem., 1995, 99, 13065 CrossRef CAS.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, Langmuir, 1996, 12, 3038 CrossRef CAS.
- (a) F. Binder, G. Calzaferri and N. Gfeller, Sol. Energy Mater. Sol. Cells, 1995, 38, 175 Search PubMed; (b) T-Q. Nguyen, J. Wu, S. H. Tolbert and B. J. Schwartz, Adv. Mater., 2001, 13, 609 CrossRef CAS.
- (a) W. Hölderlich, G. Lauth, G. Wagenblast, B. Albert, G. Lamm, H. Reichelt, C. Grund and S. Gruettner-Merten, Ger. Pat., DE 4207745, assigned to W. Hölderlich, 1992. Search PubMed; (b) W. Hölderlich, G. Lauth, U. Müller and S. Brode, Ger. Pat., DE 4131447, assigned to BASF, 1992. Search PubMed; (c) W. Hölderlich, G. Lauth, G. Wagenblast and E. Schefcizk, Ger. Pat., DE 4207339, assigned to W. Hölderlich, 1992 Search PubMed.
- S. Forrest, P. Burrows and M. Thompson, IEEE Spectrum, 2000, 38, 29 CrossRef.
- D. Wöhrle and D. Meissner, Adv. Mater., 1991, 3, 129 CrossRef CAS.
- J. Rostalski and D. Meissner, Sol. Energy Mater. Sol. Cells, 2000, 63, 37 CrossRef CAS.
- J. Wienke, T. J. Schaafsma and A. Goossens, J. Phys. Chem. B, 1999, 103, 2702 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- D. Cahen, G. Hodes, M. Grätzel, J. F. Guillemoles and I. Riess, J. Phys. Chem. B, 2000, 104, 2053 CrossRef CAS.
- R. B. M. Koehorst, G. K. Boschloo, T. J. Savenije, A. Goossens and T. J. Schaafsma, J. Phys. Chem. B., 2000, 104, 2371 CrossRef CAS.
- B. van der Zanden and A. Goossens, J. Phys. Chem. B, 2000, 104, 7171 CrossRef CAS.
- (a) D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS; (b) D. L. Dexter, J. Lumin., 1979, 18/19, 779 CrossRef.
- F. Farzad, D. W. Thompson, C. A. Kelly and G. J. Meyer, J. Am. Chem. Soc., 1999, 121, 5577 CrossRef CAS.
- T. Hayashi, T. G. Castner and R. W. Boyd, Chem. Phys. Lett., 1983, 94, 461 CrossRef CAS.
- J. L. Gole, J. A. DeVincentis and L. Seals, J. Phys. Chem. B., 1999, 103, 979 CrossRef CAS.
- C. A. Bignozzi, R. Argazzi, J. R. Schoonover, G. J. Meyer and F. Scandola, Sol. Energy Mater. Sol. Cells, 1995, 38, 187 CrossRef CAS.
- C. A. Bignozzi, J. R. Schoonover and F. Scandola, Prog. Inorg. Chem., 1997, 44, 1 CAS.
- H. Tributsch, in Proc. IPS-10, ed. Z. W. Tian and Y. Cao, International Academic Publishers, Beijing, 1993, pp. 235–247 Search PubMed.
- R. Memming and H. Tributsch, J. Phys. Chem., 1971, 75, 562 CrossRef CAS.
- H. R. Kerp, H. Donker, R. B. M. Koehorst, T. J. Schaafsma and E. E. van Faasen, Chem. Phys. Lett., 1998, 298, 302 CrossRef CAS.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265 CrossRef CAS.
- (a) I. D. W. Samuel and A. Beeby, Nature, 2000, 403, 710 CrossRef CAS; (b) V. Cleave, G. Yahioglu, P. Le Barny, R. H. Friend and N. Tessler, Adv. Mater., 1999, 11, 285 CrossRef CAS; (c) A. R. Brown, K. Pichler, N. C. Greenham, D. D. C. Bradley and R. H. Friend, Chem. Phys. Lett., 1993, 210, 61 CrossRef CAS; (d) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- J. V. Mello and N. S. Finney, Angew. Chem., 2001, 113, 1584 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |