DOI:
10.1039/B106010B
(Paper)
J. Mater. Chem., 2002,
12, 27-36
Highly-charged organic nanoparticles: redox-active dendrimers incorporating 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene units†
Received 6th July 2001, Accepted 10th October 2001
First published on 14th November 2001
Abstract
The incorporation of 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene units into dendritic structures has been achieved by convergent routes. The key starting material is the 4-(hydroxymethyl) derivative 3. The branches contain aryl ester and aryl ether groups and the core reagent is benzene-1,3,5-tricarbonyl chloride. Dendrimers 10, 15 and 18 with 6 and 12 redox units at the periphery have been characterised by 1H NMR spectroscopy, elemental analysis, mass spectrometry, UV-vis spectroscopy and solution electrochemistry. The 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene redox chemistry is retained in the dendrimer structures: clean formation of hexa(dication) and dodeca(dication) species is observed upon electrochemical oxidation. These
highly-charged species enjoy remarkable electrochemical stability as their reduction to the neutral species is thwarted by the loss of aromaticity and the marked conformational change which accompanies this process. Molecular modelling studies were performed on 10, 1012+, 18 and 1824+ by molecular mechanics.
Introduction
The study of structurally-ordered monodisperse oligomers as new materials which possess interesting optoelectronic properties is of contemporary interest.1 In this context dendrimers and related hyper-branched macromolecules continue to attract considerable attention by virtue of their well-defined, three-dimensional frameworks in which functional groups can be placed in predetermined positions thereby imparting special properties to the molecules.2 From this viewpoint, redox-active organic and organometallic groups have been built into the structures with several long-term aims in mind,3 such as (i) new electron-transfer catalysts; (ii) studies on the dynamics of electron transport at surfaces and within restricted reaction spaces; (iii) new materials for energy conversion; (iv) organic semiconductors; (v) organic magnets; and (vi) mimics
of biological redox processes.Dendrimers have been synthesised which contain a redox-active unit at the core [e.g. metalloporphyrin,4 ruthenium(II)bis(terpyridyl) complex,5 ferrocene,6 C60,7 an iron–sulfur cluster8 or bipyridinium9] and some of these display solution redox behaviour which is modulated by the outer layers of the structure hindering access of the redox site to the electrode surface.10 Alternatively, the redox units, e.g. ferrocene and related metal sandwiches,11 metal(bipyridyl),12 fullerenes,13 naphthalene diimides,14 tetrathiafulvalenes15 or
phenothiazines16 are situated at peripheral sites and/or within the branches. These redox groups may behave independently in multi-electron processes (n identical electroactive centres giving rise to a single n-electron wave at the same potential as the monomer unit) or they may interact intra- or inter-molecularly, giving rise to overlapping or closely-spaced redox waves at different potentials.
Consider the 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene system. The parent compound 117 and its derivatives, e.g.2 and 3,18 undergo a single, quasi-reversible, two-electron oxidation wave to yield a thermodynamically stable dication at Eoxca.
+0.4 V (in MeCN vs. Ag/AgCl) in the CV.19 X-Ray crystal structures have shown that a major structural change accompanies this oxidation to the dication: the central anthracenediylidene ring which is boat shaped in the neutral molecule, becomes a planar and fully aromatic anthracene system, with the 6π 1,3-dithiolium cations almost orthogonal to this plane.20 The extended π-conjugation between the
dithiolium rings and the twisted structure serves to remove intramolecular Coulombic repulsion.21System 1 is, therefore, very different from those redox species mentioned above, for which there is very little structural change upon oxidation or reduction. Theoretical calculations have deduced that the saddle-shape of neutral 1 arises from steric hindrance between the sulfur atoms and the hydrogen atoms at the peri sites of the anthracenediylidene system, and this is completely relieved in the dication.22 This interplay of unusual structural and redox properties has led to the use of monomeric derivatives of 1 as components of intermolecular20,23 and intramolecular24 charge-transfer systems.
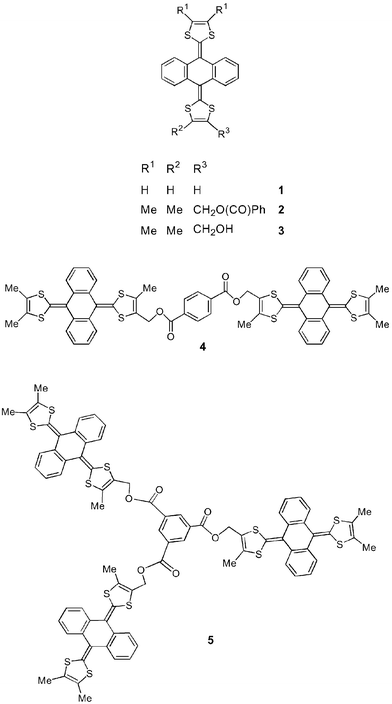
We now present the first incorporation of 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene units into dendritic structures using convergent methodology and discuss their UV-vis spectroscopic, redox and structural properties. At the outset we reasoned that highly-charged redox states should be attainable, and with dense surface packing intradendrimer interactions might be manifest. Prior to this work only a very few dimers, e.g.4,18,25 and one trimer 518 of system 1 were known.
Results and discussion
Synthesis
Recent synthetic developments have made 3 readily available in 2–3 g batches,18 and we established that aryl ester derivatives could be obtained under mild conditions, e.g. reaction with 1,3,5-benzenetricarbonyl chloride (9) afforded trimer 5 in 78% yield.18 We, therefore, considered 3 to be a convenient starting reagent for a convergent synthesis of dendritic structures.The reaction of 3 with the silyl-protected diacid chloride 6,26 using triethylamine as base in dichloromethane at room temperature, gave 7
(97% yield), deprotection of which [tetra-n-butylammonium fluoride (TBAF) in tetrahydrofuran (THF) at room temperature] afforded the dendron wedge 8
(99% yield) containing the phenolic group as a reactive handle for further functionalisation. By analogy with the preparation of 7, the hexakis[anthracenebis(dithiole)] derivative 10 was obtained in 77% yield from reaction of 8 with 9
(Scheme 1). Similarly, reaction of 8 with reagent 6 gave the protected second generation wedge 11
(72% yield). Repeated attempts at desilylation of 11 unexpectedly led to fragmentation of the molecule and essentially quantitative conversion back to 8. We were, therefore, unable to synthesise higher generation analogues of 10.
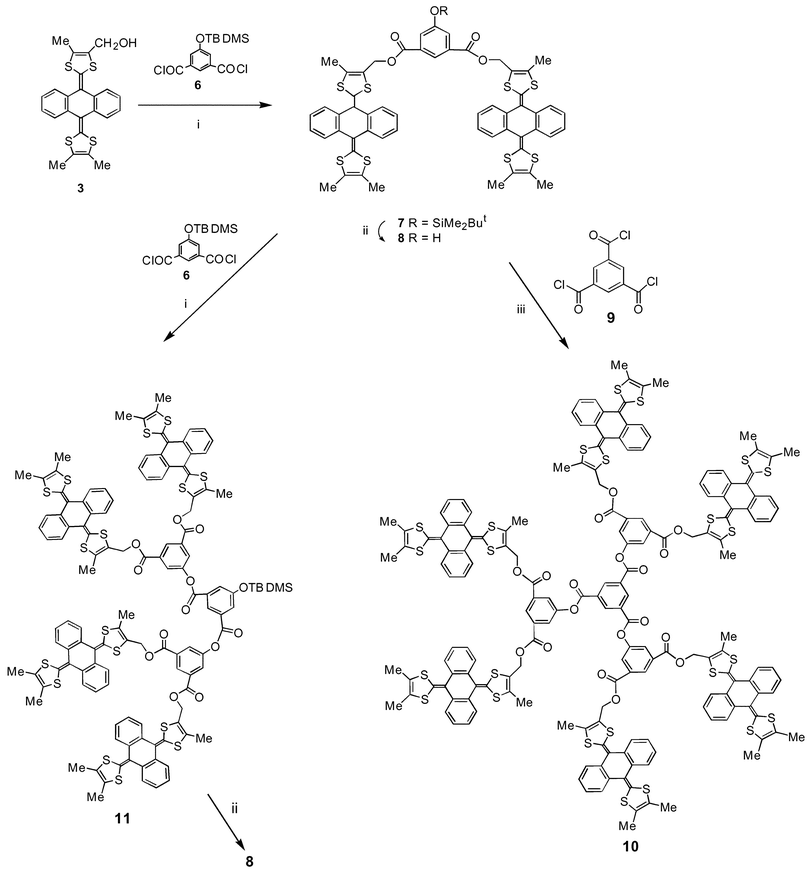 |
| | Scheme 1 Reagents and conditions: i, Et3N, CH2Cl2, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ºC; ii, TBAF, THF, 20ºC; iii, Et3N, CH2Cl2, 20ºC. ºC; ii, TBAF, THF, 20ºC; iii, Et3N, CH2Cl2, 20ºC. | |
To circumvent this problem we incorporated more robust ether linkages into the dendritic scaffold, using the strategy developed by Fréchet for poly(arylether) systems.27 Compound 3 was converted into the chloromethyl derivative 12 in 95% yield by treatment with triphenylphosphine–carbon tetrachloride. Two-fold reaction of 12 with 3,5-dihydroxybenzyl alcohol 13, using potassium carbonate as base, afforded 14, albeit in only 28% yield. The disappointing yield for this reaction is due to the heterogeneous mixture and poor reactivity of 12. Compound 14 was smoothly converted into 15
(57%)
and 16
(65%), by direct analogy with the preparation of 10 and 11. Unlike 11, 16 was cleanly deprotected (TBAF, THF) to yield alcohol derivative 17
(70% yield)
(Scheme 2). Three-fold esterification of 17 with core reagent 9 gave the dodeca[anthracenebis(dithiole)] derivative 18
(54% yield)
(Scheme 3). Dendrimers 10, 15 and 18
were isolated as air-stable orange solids and their structures were unambiguously established by a combination of 1H NMR spectroscopy, mass spectrometry and elemental analysis. The 1H NMR spectra of 10 and 18 are shown in Figs. 1 and 2, respectively.
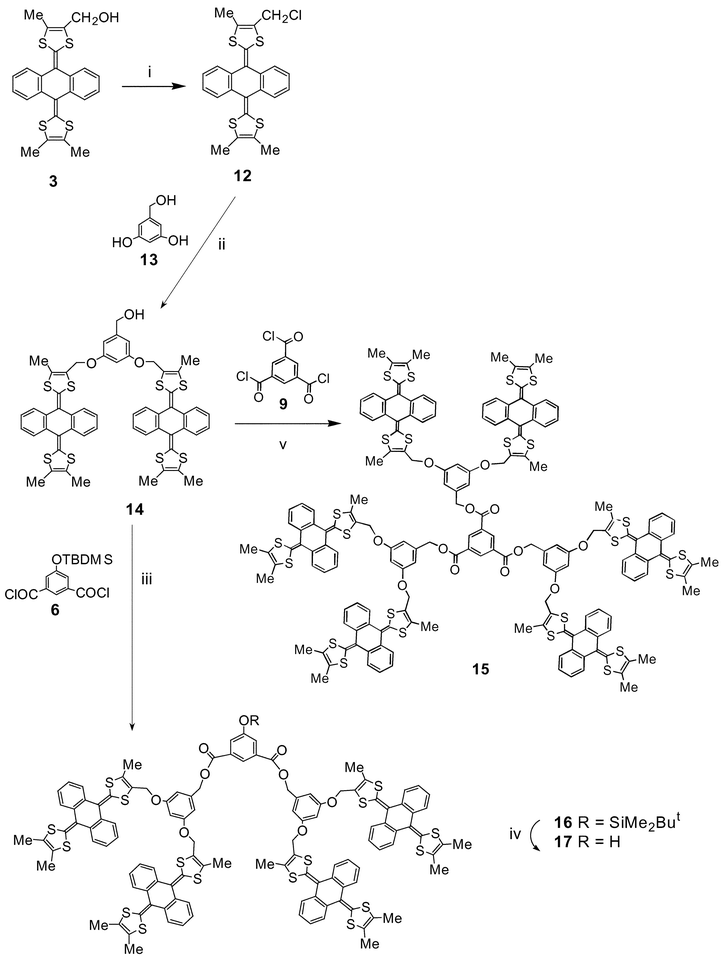 |
| | Scheme 2 Reagents and conditions: i, PPh3, CCl4, MeCN, reflux; ii, K2CO3, 18-crown-6, THF, reflux; iii, Et3N, CH2Cl2, 20ºC; iv, TBAF, THF, 20ºC; v, Et3N, CH2Cl2, 20ºC. | |
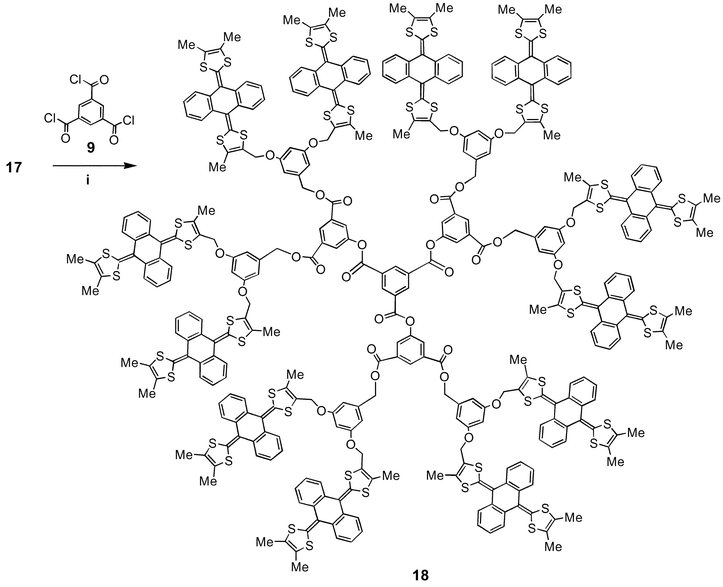 |
| | Scheme 3 Reagents and conditions: i, Et3N, CH2Cl2, 20ºC. | |
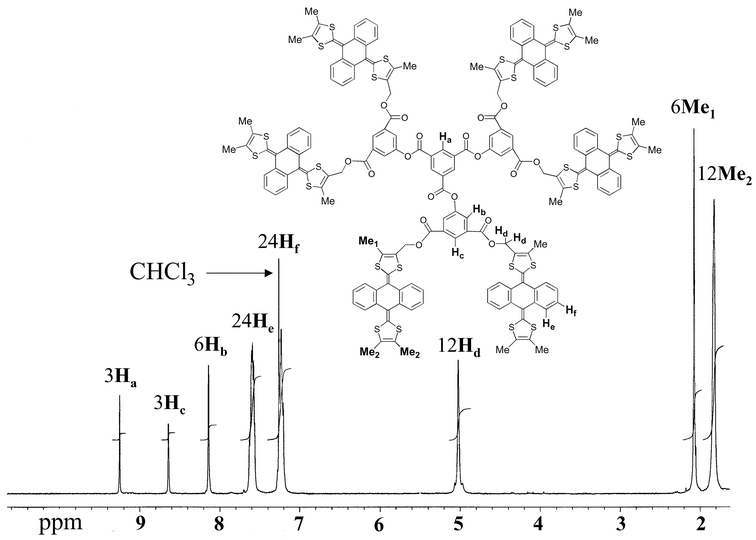 |
| | Fig. 1 1H NMR spectrum of 10 in CDCl3. | |
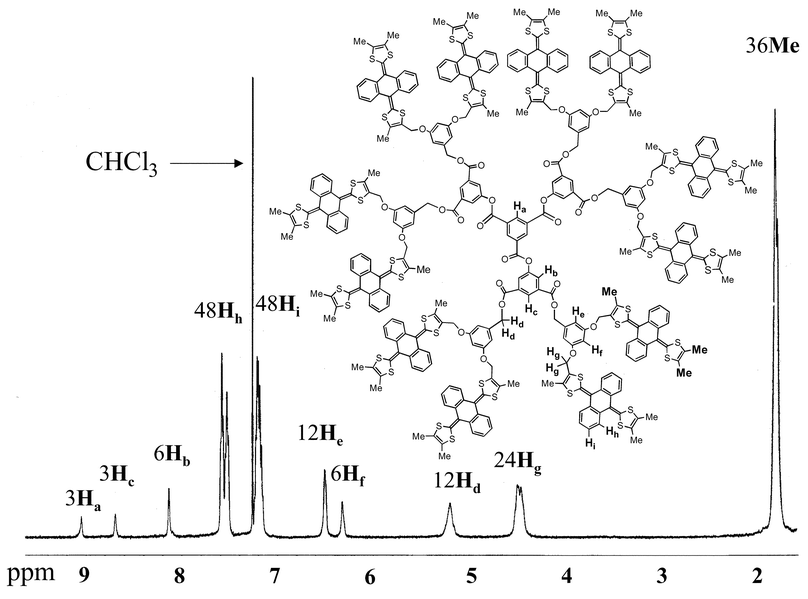 |
| | Fig. 2 1H NMR spectrum of 18 in CDCl3. | |
Solution electrochemistry
Solution electrochemical data, obtained by cyclic voltammetry (CV) are collated in Table 1. The benzoyl ester 2 shows the typical quasireversible two-electron redox wave (neutral → dication species) observed for 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene derivatives.19,20b A comparison of the model compound 2 with the dendritic molecules 5, 10, 15 and 18 reveals some interesting trends. The oxidation potential (Eoxpa) is almost identical for all the compounds [ca. 500 mV in dichloromethane–acetonitrile (2∶1 v/v) and 600 mV
in THF (Figs. 3 and 4)]. All the peripheral redox units are independent and oxidised at the same time. A slight broadening of the oxidation peak can be noticed with increasing generation. This feature has already been reported with certain other dendrimers containing redox moieties at the periphery.3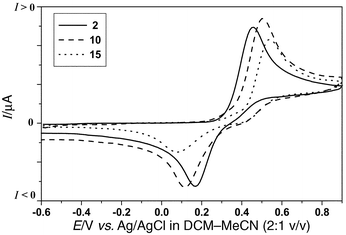 |
| | Fig. 3 Cyclic voltammograms of 2, 10 and 15 under the conditions stated in Table 1. | |
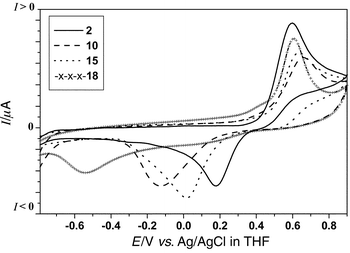 |
| | Fig. 4 Cyclic voltammograms of 2, 10, 15 and 18 under the conditions stated in Table 1. | |
Table 1 Cyclic voltammetric dataa
| Compound | THF | DCM–acetonitrile (2∶1 v/v) |
|---|
| Eox/V | Ered/V | ΔE/Vb | Eox/V | Ered/V | ΔE/Vb |
|---|
| Versus Ag/AgCl, electrolyte Bu4N+PF6−, 20°C, scan rate 100 mVs−1. ΔE
=
Eox − Ered. Not determined. |
|---|
| 2 | 0.60 (2e) | 0.18 | 0.42 | 0.46 (2e) | 0.16 | 0.30 |
| 5 | 0.62 (6e) | 0.16 | 0.46 | n.d.c | n.d.c | n.d.c |
| 10 | 0.66 (12e) | −0.13 | 0.79 | 0.51 (12e) | 0.11 | 0.40 |
| 15 | 0.64 (12e) | 0.02 | 0.62 | 0.54 (12e) | 0.08 | 0.46 |
| 18 | 0.61 (24e) | −0.54 | 1.15 | Not soluble | | |
A striking feature of these data is that as the generation increases there is a decrease in the reversibility of the redox wave in THF, leading to a complete electrochemical irreversibility for 18
(ΔE = 1150 mV). However, in a mixture of DCM–MeCN, where the charged species are more soluble, this irreversibility is less pronounced. This is consistent with precipitation of the charged species occurring on the surface of the Pt electrode in THF, which does not take place in DCM–MeCN. This was confirmed by the electrode being completely blocked after a few scans around Eoxpa for 10 and 18 in THF which prevents their use as modified electrodes, cf. the ferrocenyl dendrimers reported recently by Astruc et al.11e and Cuadrado et
al.11d which are useful in this regard. Consequently, the decrease of reversibility for 10 and 15 in DCM–MeCN (ΔE increased by 100 mV and 160 mV, respectively) can be attributed to the dramatic change of conformation which must occur on reduction of the oxidised forms of 1012+ and 1512+ back to their neutral forms. This represents an interesting dendritic effect on the solution redox chemistry.
Chemical oxidation and reduction
Addition of iodine to a dichloromethane solution of 10, 15 and 18 gave red–purple precipitates of the iodide salts of 10n+, 15n+ and 18n+. These precipitates could be reduced back to the neutral species by adding sodium hydrosulfite and heating the mixture. The 1H NMR spectra recorded before oxidation and after reduction were identical, showing that the chemical redox processes in these dendrimers are reversible without any significant decomposition of the oxidised species. Evidence for complete oxidation to 1012+ was provided by 1H NMR data in dry DMF in which both the neutral
and oxidised species are soluble: in particular, the methyl and CH2 protons (shown as Me1, Me2 and Hd in Fig. 1) shifted downfield from δ 2.19, 1.88 and 5.23 to δ 3.36, 3.10 and 6.28, respectively, consistent with each unit being oxidised to the bis(1,3-dithiolium) cation, as shown previously in monomeric derivatives.28 No paramagnetic broadening of the NMR spectra was observed, confirming that no cation radical species derived from 10 were present.UV-Vis spectroscopy
UV-Vis spectra were obtained in DCM solution. The benzoyl ester 2 served as a model compound, and showed the two characteristic absorption bands at λmax = 368 and 436 nm for the 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene system.20c The spectra are shown in Fig. 5. UV-Vis spectra of 2 were recorded for a range of concentrations varying from C to 20 × C
(where C = 6.31 × 10−6 M). The absorbances at λmax = 368 and 436 nm vary as a linear function of the concentration (Fig. 5 inset). UV-Vis spectra of 10, 15 and 18 were recorded at the same concentration C = 6.31 × 10−6 M. The spectra obtained for 10 and 15
(curves II and III, respectively) are equivalent to a 6 × C spectrum of 2, and for 18
(curve I) equivalent to a 12 × C spectrum of 2. Consequently, the extinction coefficient of each dendrimer corresponds to the extinction coefficient of one chromophore unit multiplied by the number of units in the dendrimer. This confirms that each chromophore unit is acting independently, with no dendritic effect observed in the optical spectra. Therefore, this spectroscopic
technique could also be used to quantify the number of units present for a new dendrimer.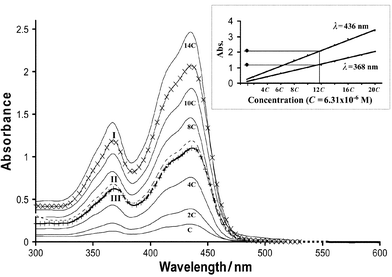 |
| | Fig. 5 UV-Vis spectra of 2 at varying concentrations C to 14 × C, 10
(concentration C, curve II), 15
(concentration C, curve III) and 18
(concentration C, curve I); C = 6.31 × 10−6 M in CH2Cl2. The inset shows the correlation of the maxima of absorption for 18 at concentrations C to 20 × C
(black circles) to the linear variation of absorbance as a function of the concentration of 2, for wavelengths λmax 368 and 436 nm. | |
Molecular modelling studies
In order to visualise the change in conformation occurring during oxidation, molecular modelling studies were performed on 10, 1012+, 18 and 1824+ by molecular mechanics. Energy minimisation of the 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene unit in the neutral state showed the most favourable saddle-shape conformation which is in agreement with previous X-ray crystal structures,20 variable temperature solution 1H NMR data18 and theoretical calculations on monomeric derivatives.22 Energy minimisation of the core was then performed before the addition of the energy-minimised units at its periphery. Geometry optimisation of the dendrimers 10
and 18 was then calculated and the resulting converged structures are shown in Figs. 6 and 7. The energy-minimised conformations of 1012+ and 1824+ were obtained by adding to the energy-minimised conformation of 10 and 18, a positive charge on the C(2) atom of each dithiole ring (i.e. 12 and 24 positive charges in total for 10 and 18, respectively). The structures obtained for 1012+ and 1824+ show that each unit adopts the characteristic dicationic shape wherein the anthracene
moiety is planar with the two dithiolium units perpendicular to it, which is in accord with theoretical calculations and X-ray crystal structures.20 It also complies with a maximum space expansion between the units arising from Coulombic repulsion between the positively charged dithiolium rings.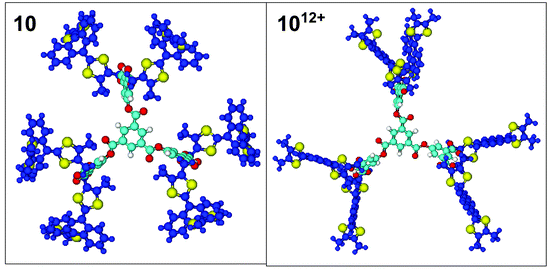 |
| | Fig. 6 Molecular model of 10
(left view) and 1012+
(right view) perpendicular to the plane of the core benzene ring. Colour code: 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene system (purple); sulfur (yellow); oxygen (red); remaining carbon scaffold (light blue). | |
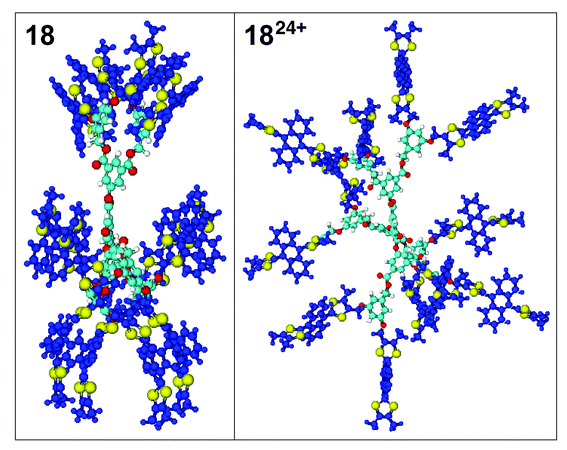 |
| | Fig. 7 Molecular model of 18
(left view) and 1824+
(right view) along the plane of the benzene ring. Colour codes are the same as Fig. 6. | |
It should be noted that for the neutral form of dendrimer 10 other unsymmetrical local minima of comparable energy can be reached where π–π interactions occur between the units. The proximity of the 12 units of dendrimer 18 leads also to π–π interactions between the units and consequently to several converged unsymmetrical structures, but also to a symmetrical one of lowest energy presented in Fig. 7. On the contrary, starting from different non-optimised geometries of 1012+ and 1824+, only one converged structure for each species was observed, in which all the charged units are as far as possible from each other. The structures shown for 10 and 18
provide a good comparison between the shape of the neutral and oxidised forms of the dendrimers. The average molecular diameter of their spherical shapes is 40 Å for 10 and 46 Å for 1012+. This is representative of the compact form of 10 imposed by the saddle-shape conformation of the units, compared to the more extended shape of 1012+ imposed by the Coulombic repulsion. In the case of dendrimer 18, this feature is even more pronounced especially due to the π–π interactions occurring inside the dendritic structure. Consequently, dendrimer 18 presents a cylindrical shape (ca. 52 Å × 30 Å) and its oxidised
form the expected spherical shape with an average molecular diameter of 61 Å. It is also noteworthy that there are cavities inside the neutral and oxidised forms that could offer some interesting host–guest properties which are currently under investigation.
Conclusions
In this work we have synthesised the first dendrimers to incorporate the anthracene-bis(dithiole) system: macromolecules with 6 and 12 of these redox units at the periphery have been obtained by convergent methodology with aryl ester and aryl ether units in the branches, joined to a 1,3,5-benzene triester core unit. These compounds possess well-defined redox activity, and the two-electron oxidation of each of the redox units affords species with a high charge: molecular weight ratio. Charged organic nanoparticles of this type are relevant to the miniaturisation of electronic materials.29 The dendritic wedges synthesised during the course of this work, e.g.8, 14 and 17, which possess reactive alcohol substituents, are available in synthetically useful quantities and should be amenable to other synthetic transformations, e.g.
to yield dendronised polymers30 or new systems which undergo multi-electron charge-transfer processes.Experimental
General methods and procedures
1H and 13C NMR spectra were obtained on Oxford 200, Varian Unity 300 and Varian VXR 400S spectrometers operating at 199.992 (1H) and 50.293 (13C), 299.908 (1H) and 75.420 (13C), and 400.0 (1H) and 100.6 (13C) MHz, respectively. Mass spectra were recorded on a Micromass Autospec spectrometer operating at 70 eV. MALDI TOF mass spectra were obtained on a Kratos IV instrument in the reflection mode, operating with irradiation from a nitrogen laser at 337 nm. The matrix was 2,5-dihydroxybenzoic acid, and spectra were averaged over 100 pulses whilst scanning across the sample: peak half-widths were between 6–10 amus. Infra-red spectra were recorded using KBr disks on a Perkin Elmer 1600 FTIR, spectrometer operated from a Grams Analyst 1600. Electronic absorption spectra were obtained using a Perkin Elmer II UV-vis spectrophotometer
operating with 1 ml quartz cells. Melting points were obtained on a Philip Harris melting point apparatus and are uncorrected. Cyclic voltammetric data were measured with iR compensation using a BAS CV50 electrochemical analyser. The experiments were carried out with 5 cm3 of a ca. 10−4 M solution of the compound in DCM containing 0.1 M tetrabutylammonium perchlorate as the electrolyte, at scan rate 100 mV s−1. The oxidation potentials (which represent a two-electron process) were measured versus a platinum wire quasi-reference electrode and corrected versus decamethylferrocene/decamethylferrocenium+ by adding decamethylferrocene to the studied solution after the experiment, and referenced versus Ag/AgCl. Structures were modeled and optimised in the gas phase without consideration of the counterions using the MM+ programme
Hyperchem 5.0 (Hyperchem, Hypercube Inc., 1115 NW 4th Street, Gainsville FL 32601, USA) following the technique of other workers.31Column chromatography was carried out using Merck silica gel (70–230 mesh) and solvents were distilled prior to use in column chromatography. All reactions were performed under an inert atmosphere of nitrogen in pre-dried glassware.
Dimer 7
To a stirred solution of alcohol 318
(800 mg, 1.77 mmol) in dry DCM (100 cm3) at 20°C was added diacid chloride 626
(320 mg, 0.885 mmol) and triethylamine (0.5 cm3, 3.54 mmol). The resultant mixture was stirred overnight. After evaporation of the solvents in vacuo, the residue was purified by chromatography eluting with DCM to afford 7
(1.00 g, 97%) as an orange solid, mp 215°C; δH
(CDCl3) 8.26 (s, 1 H), 7.6 (m, 10 H), 7.2 (m, 8 H), 5.01 (s, 4 H), 2.09 (s, 6 H), 1.88 (s, 12 H), 0.98 (s, 9 H), 0.20 (s, 6 H); νmax
(KBr)
1727, 1521, 1445, 1219, 755, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2) 368 (4.33), 436 (4.58); m/z
(MALDI-TOF) 1164 (M+); [Analysis found: C, 63.76; H, 5.01; C62H56O5S8Si (1164.17) requires: C, 63.88; H 4.84%].Dimer 8
To a stirred solution of 7
(200 mg, 0.17 mmol) in dry THF (50 cm3) at 20°C was added tetrabutylammonium fluoride (1 M in THF, 0.17 cm3, 0.17 mmol). The resultant mixture was stirred for 1 h. The solvents were removed by evaporation in vacuo and the residue was purified by chromatography with DCM containing 2% of MeOH as eluent, to afford 8
(178 mg, 99%) as an orange solid, mp > 250°C; δH
(CDCl3) 8.24 (s, 1 H), 7.6 (m, 10 H), 7.2 (m, 8 H), 5.01 (s, 4 H), 2.09 (s, 6 H), 1.89 (s, 12 H); νmax
(KBr) 3500 (br), 1727, 1521, 1445, 1219, 755, 675, 645 cm−1; λmax
(lg ε)
(CH2Cl2)
368 (4.39), 436 (4.63); m/z
(MALDI-TOF) 1050 (M+); [Analysis found: C, 63.72; H, 4.04; C56H42O5S8
(1051.46) requires: C, 63.97; H, 4.03%].Dendrimer 10
By analogy with the preparation of 7, alcohol 8
(100 mg, 0.1 mmol), 1,3,5-benzenetricarbonyl trichloride (9) (8.4 mg, 0.033 mmol) and triethylamine (26 µl, 0.2 mmol) in dry DCM (20 cm3) were stirred at 20°C. Chromatography eluting with DCM afforded 10
(81 mg, 77%) as an orange solid, mp > 250°C; δH
(CDCl3) 9.26 (s, 3 H), 8.64 (s, 3 H), 8.14 (s, 6 H), 7.6 (m, 24 H), 7.2 (m, 24 H), 5.03 (s, 12 H), 2.08 (s, 18 H), 1.83 (s, 36 H); νmax
(KBr) 1729, 1520, 1444, 1213, 754, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2)
368 (5.03), 436 (5.27); m/z
(MALDI-TOF) 3308 (M+); [Analysis found: C 63.81, H 4.05; C177H126O18S24, (3310.47) requires: C, 64.22; H 3.84%].Dendron wedge 11
Following the same procedure as 7, alcohol 8
(550 mg, 0.52 mmol), diacid chloride 6
(100 mg, 0.26 mmol) and triethylamine (0.14 cm3, 1.04 mmol) afforded 11
(450 mg, 72%) as an orange solid, mp 230°C (decomp.: black powder at >200°C); δH
(CDCl3) 8.62 (m, 3 H), 8.11 (d, J 1.4, 4 H), 7.89 (d, J 1.2, 4 H), 7.6 (m, 16 H), 7.2 (m, 16 H), 5.02 (s, 8 H), 2.09 (s, 12 H), 1.85 (s, 24 H), 1.03 (s, 9 H), 0.29 (s, 6 H); νmax
(KBr) 1728, 1519, 1444, 1215, 756, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2)
368 (4.85), 436 (5.12); m/z
(MALDI-TOF) 2362 (M+); C126H100O13S16Si requires 2363.28.10-(4,5-Dimethyl-1,3-dithiol-2-ylidene)-9,10-dihydro-9-(4-chloromethyl-5-methyl-1,3-dithiol-2-ylidene)anthracene 12
To a stirred solution of 3
(2.00 g, 4.4 mmol) in dry acetonitrile (50 cm3) was added triphenylphosphine (1.25 g, 4.8 mmol) and carbon tetrachloride (20 cm3). After 1 h at reflux the colour changed from orange to dark brown and TLC monitoring showed the reaction was complete. The mixture was filtered through a plug of silica gel, eluting with EtOAc and then concentrated in vacuo. Flash chromatography of the residue eluting with EtOAc–hexane (1∶2 v/v) afforded 12
(2.00 g, 95%) as an orange solid, mp 225°C (decomp. to a black powder); δH
(CDCl3) 7.66–7.59 (m, 4 H), 7.29–7.25 (m, 4 H), 4.27 (s, 2 H), 2.03 (s, 3 H), 1.93 (s,
6 H); νmax
(KBr) 1512, 1456, 1445, 755, 675, 644 cm−1; λmax
(lg ε)
(EtOAc) 364 (4.24), 428 (4.49). [Analysis found: C, 61.26; H, 4.13; C24H19ClS4
(471.12) requires C, 61.18; H, 4.06%].(N.B. This compound is easily decomposed on silica and especially in solvents like DCM, or in acid conditions).
Dendron wedge 14
To a stirred solution of 12
(2.00 g, 4.2 mmol) in dry THF (50 cm3) was added 3,5-dihydroxybenzyl alcohol (298 mg, 2.1 mmol), K2CO3
(640 mg, 4.2 mmol) and 18-crown-6 (112 mg, 0.4 mmol). The resulting mixture was heated at reflux and vigorously stirred for 24 h. The solvent was evaporated in vacuo. Water (100 cm3) was added to the residue and the crude product extracted with chloroform (3 × 50 cm3). The combined organic layers were dried over MgSO4 and evaporated to dryness. The residue was chromatographed (eluent: dichloromethane) to afford 14
(600 mg, 28%) as an orange solid, mp ca. 240 °C; δH
(CDCl3)
7.65–7.61 (m, 8 H), 7.28–7.23 (m, 8 H), 6.56 (s, 2 H), 6.42 (s, 1 H). 4.64 (s, 4 H), 4.61 (s, 2 H). 1.98 (s, 6 H), 1.90 (s, 12 H); νmax
(KBr) 3450 (br) 1595, 1520, 1456, 1444, 1281, 1144, 755, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2) 368 (4.56), 436 (4.80); m/z
(MALDI-TOF) 1009 (M+); [Analysis found: C, 64.77; H, 4.35; C55H44O3S8
(1009.5)
(+0.5 mol of H2O) requires: C, 64.86; H 4.45%].Dendrimer 15
By analogy with the preparation of 7, alcohol 14
(100 mg, 0.1 mmol), 1,3,5-benzenetricarbonyl trichloride (9) (8.4 mg, 0.03 mmol) and triethylamine (26 µl, 0.2 mmol) in dry DCM (10 cm3) were stirred at room temperature. Chromatography on silica gel eluting with dichloromethane afforded dendrimer 15
(60 mg, 57%) as an orange solid, mp 245°C, δH
(CDCl3) 8.92 (s, 3 H), 7.62–7.54 (m, 24 H), 7.26–7.21 (m, 24 H), 6.52 (s, 6 H), 6.37 (s, 3 H), 5.22 (s, 6 H), 4.51 (s, 12 H), 1.84 (br s, 54 H); λmax
(lg ε)
(CH2Cl2)
368 (4.99), 436 (5.24); m/z
(MALDI-TOF) 3184 (M+); [Analysis found: C, 65.42; H, 4.03; C174H132O12S24
(3184.49) requires C, 65.63; H 4.18%].Dendron wedge 16
Following the same procedure as for 7, alcohol 14
(300 mg, 0.3 mmol), diacid chloride 6
(54 mg, 0.15 mmol) and triethylamine (80 µL, 0.6 mmol) afforded 16
(220 mg, 65%) as an orange solid, mp ca. 245°C; δH
(CDCl3) 8.34 (br s, 1 H), 7.73 (s, 2 H), 7.65–7.54 (m, 16 H), 7.29–7.22 (m, 16 H), 6.54 (br s, 4 H), 6.37 (br s, 2 H), 5.20 (br s, 4 H), 4.51 (br s, 8 H), 1.84 (br s, 36 H), 0.99 (s, 9 H), 0.23 (s, 6 H); νmax
(KBr) 1725, 1595, 1520, 1444, 1216, 1144, 754, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2)
368 (4.81), 436 (5.03); m/z
(MALDI-TOF) 2278 (M+); [Analysis found: C, 65.43; H, 4.73; C124H104O9S16Si (2279.3) requires C, 65.34; H, 4.60%].Dendron wedge 17
Following the same procedure as for 8, dendron 16
(200 mg, 0.09 mmol) was dissolved in dry THF (50 cm3). Tetrabutylammonium fluoride (1 M in THF, 0.09 cm3, 0.09 mmol) was added and the resultant mixture was stirred for 1 h. The solvent was removed by evaporation in vacuo and the residue was purified by chromatography with DCM as eluent, to afford 17
(133 mg, 70%) as an orange solid, mp 245°C (decomp.); δH
(CDCl3) 8.30 (br s, 1 H), 7.71 (s, 2 H), 7.65–7.54 (m, 16 H), 7.27–7.17 (m, 16 H), 6.52 (br s, 4 H), 6.37 (br s, 2 H), 5.20 (br s, 4 H), 4.52 (br s, 8 H) 1.84 (br
s, 36 H); νmax
(KBr) 3450 (br), 1726, 1596, 1520, 1445, 1222, 1146, 755, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2) 368 (4.85), 436 (5.09); m/z
(MALDI-TOF) 2164 (M+); [Analysis found: C, 64.15; H, 4.23; C118H90O9S16
(2165.0)
(+2 mol of H2O) requires: C, 64.39; H 4.30%].Dendrimer 18
By analogy with the preparation of 7, alcohol 17
(130 mg, 0.06 mmol), 1,3,5-benzenetricarbonyl trichloride (9) (5.3 mg, 0.02 mmol) and triethylamine (16 µl, 0.12 mmol) in dry DCM (10 cm3) were stirred at 20°C. Chromatography eluting with DCM afforded 18
(72 mg, 54%) as an orange solid, mp 240°C (decomp.); δH
(CDCl3) 9.05 (s, 3 H), 8.69 (s, 3 H), 8.14 (s, 6 H), 7.59–7.46 (m, 48 H), 7.28–7.18 (m, 48 H), 6.51 (s, 12 H), 6.33 (s, 6 H), 5.22 (br s, 12 H), 4.50 (m, 24 H), 1.82–1.79 (br s, 108 H); νmax
(KBr)
1731, 1596, 1520, 1444, 1222, 1146, 753, 675, 644 cm−1; λmax
(lg ε)
(CH2Cl2) 368 (5.27), 436 (5.51); m/z
(MALDI-TOF) 6648 (M+); [Analysis found: C, 65.37; H, 4.23; C363H270O30S48
(6651.2) requires: C 65.55, H 4.09%].Acknowledgements
We thank the University of Durham for funding.References
-
(a) Reviews: Electronic Materials: The Oligomer Approach, Eds. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1998 Search PubMed;
(b) R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350 CrossRef;
(c) Y. Shirota, J. Mater. Chem., 2000, 10, 1 RSC;
(d) D. Fichou, J. Mater. Chem., 2000, 10, 571 RSC;
(e) J. L. Segura and N. Martín, J. Mater. Chem., 2000, 10, 2403 RSC.
-
(a) Reviews: D. A. Tomalia, A. M. Naylor and W. A. Goddard, III, Angew. Chem., Int. Ed. Engl., 1990, 29, 138 Search PubMed;
(b) G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendritic
Molecules: Concepts, Synthesis, Perspectives, VCH, Weinheim, 1996 Search PubMed;
(c) O. A. Matthews, A. N. Shipway and J. F. Stoddart, Prog. Polym. Sci., 1998, 23, 1 CrossRef;
(d) M. Fischer and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 884 CrossRef;
(e) S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS;
(f) Dendrimers II: Architecture, Nanostructure and Supramolecular Chemistry, Ed. F. Vögtle,Springer, London, 2000 Search PubMed.
- For a review of redox-active dendrimers see: M. R. Bryce and W. Devonport, in Advances in Dendritic Macromolecules, Ed. G. R. Newkome, JAI Press, London, 1996, Volume 3, p. 115 Search PubMed.
- P. Weyermann, J.-P. Gisselbrecht, C. Boudon, F. Diederich and M. Gross, Angew. Chem., Int. Ed., 1999, 38, 3215 CrossRef CAS.
- M. Kimura, T. Shiba, T. Muto, K. Hanabusa and H. Shirai, Chem. Commun., 2000, 11 RSC.
- C. M. Cardona and A. E. Kaifer, J. Am. Chem. Soc., 1998, 120, 4023 CrossRef CAS.
- X. Camps, E. Dietel, A. Hirsch, S. Pyo, L. Echegoyen, S. Hackbarth and B. Röder, Chem. Eur. J., 1999, 5, 2362 CrossRef CAS.
- C. B. Gorman, J. C. Smith, M. W. Hager, B. L. Parkhurst, H. Sierzputowska-Gracz and C. A. Haney, J. Am. Chem. Soc., 1999, 121, 9958 CrossRef CAS.
- R. Toba, J. M. Quintela, C. Peinador, E. Román and A. E. Kaifer, Chem. Commun., 2001, 857 RSC.
- For a review of encapsulated redox centres in dendrimers and other structures, see: C. M. Cardona, S. Mendoza and A. E. Kaifer, Chem. Soc. Rev., 2000, 29, 37 Search PubMed.
-
(a) C. Valério, E. Alonso, J. Ruiz, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 1999, 38, 1747 CrossRef CAS;
(b) Review: M. A. Hearshaw and J. R. Moss, Chem. Commun., 1999, 1 Search PubMed;
(c) S. Nlate, J. Ruiz, J.-C. Blais and D. Astruc, Chem. Commun., 2000, 417 RSC;
(d) S. Nlate, J. Ruiz, V. Sartor, R. Navarro, J.-C. Blais and D. Astruc, Chem. Eur. J., 2000, 6, 2544 CrossRef CAS;
(e) Review: C. M. Casado, I. Cuadrado, M. Morán, B. Alonso, B. García, B. González and J. Losada, Coord. Chem. Rev., 1999, 185–186, 53 Search PubMed.
-
(a) S. Serroni, A. Juris, M. Venturi, S. Campagna, I. R. Resino, G. Denti, A. Credi and V. Balzani, J. Mater. Chem., 1997, 7, 1227 RSC;
(b) G. R. Newkome, A. K. Patri and L. A. Godínez, Chem. Eur. J., 1999, 5, 1445 CrossRef CAS.
- N. Armaroli, C. Boudon, D. Felder, J.-P. Gisselbrecht, M. Gross, G. Marconi, J.-F. Nicoud, J.-F. Nierengarten and V. Vicinelli, Angew. Chem., Int. Ed., 1999, 38, 3731 CrossRef.
- I. Tabakovic, L. L. Miller, R. G. Duan, D. C. Tully and D. A. Tomalia, Chem. Mater., 1997, 9, 736 CrossRef CAS.
-
(a) M. R. Bryce, W. Devonport, L. M. Goldenberg and C. Wang, Chem. Commun., 1998, 945 RSC;
(b) M. R. Bryce, J. Mater. Chem., 2000, 10, 589 RSC;
(c) C. A. Christensen, M. R. Bryce and J. Becher, Synthesis, 2000, 1695 CrossRef CAS.
- J. B. Christensen, M. F. Nielsen, J. A. E. H. van Haare, M. W. P. L. Baars, R. A. J. Janssen and E. W. Meijer, Eur. J. Org. Chem., 2001, 2123 CrossRef CAS.
- A. J. Moore and M. R. Bryce, J. Chem. Soc., Perkin Trans. 1, 1991, 157 RSC.
- N. Godbert, M. R. Bryce, S. Dahaoui, A. S. Batsanov, J. A. K. Howard and P. Hazendonk, Eur. J. Org. Chem., 2001, 749 CrossRef CAS.
-
(a) For discussions of redox processes
in derivatives of the title system see: M. R. Bryce, M. A. Coffin, M. B. Hursthouse, A. I. Karaulov, K. Müllen and H. Scheich, Tetrahedron Lett., 1991, 32, 6029 Search PubMed;
(b) S.-G. Liu, I. Pérez, N. Martín and L. Echegoyen, J. Org. Chem., 2000, 65, 9092 CrossRef CAS.
-
(a) M. R. Bryce, A. J. Moore, M. Hasan, G. J. Ashwell, A. T. Fraser, W. Clegg, M. B. Hursthouse and A. I. Karaulov, Angew. Chem., Int. Ed. Engl., 1990, 29, 1450 CrossRef;
(b) M. R. Bryce, T. Finn, A. S. Batsanov, R. Kataky, J. A. K. Howard and S. B. Lyubchik, Eur. J. Org. Chem., 2000, 1199 CrossRef CAS;
(c) A. E. Jones, C. A. Christensen, D. F. Perepichka, A. S. Batsanov, A. Beeby, P. J. Low, M. R. Bryce and A. W. Parker, Chem. Eur. J., 2001, 7, 973 CrossRef CAS.
-
(a) For discussions of Coulombic effects in multi-stage redox systems
see: K. Deuchert and S. Hünig, Angew. Chem., Int. Ed. Engl., 1978, 17, 875 Search PubMed;
(b) S. Hünig and H. Berneth, Top. Curr. Chem., 1980, 92, 1 Search PubMed.
- N. Martín, L. Sánchez, C. Seoane, E. Ortí, P. M. Viruela and R. Viruela, J. Org. Chem., 1998, 63, 1268 CrossRef CAS.
- S. Triki, L. Ouahab, D. Lorcy and A. Robert, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, C49, 1189 CrossRef.
-
(a) M. A. Herranz, N. Martín, L. Sánchez, J. Garín, J. Orduna, R. Alcalá, B. Villacampa and C. Sánchez, Tetrahedron, 1998, 54, 11651 CrossRef CAS;
(b) M. A. Herranz and N. Martín, Org. Lett., 1999, 1, 2005 CrossRef CAS;
(c) C. A. Christensen, M. R. Bryce, A. S. Batsanov, J. A. K. Howard, J. O. Jeppesen and J. Becher, Chem. Commun., 1999, 2433 RSC;
(d) N. Martín, L. Sánchez and D. M. Guldi, Chem. Commun., 2000, 113 RSC.
- N. Martín, I. Pérez, L. Sánchez and C. Seoane, J. Org. Chem., 1997, 62, 870 CrossRef CAS.
- T. M. Miller, E. W. Kwok and T. X. Neenan, Macromolecules, 1992, 25, 3143 CrossRef CAS.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- N. Godbert, A. S. Batsanov, M. R. Bryce and J. A. K. Howard, J. Org. Chem., 2001, 66, 713 CrossRef CAS.
- U. Simon, Adv. Mater., 1998, 10, 1487 CrossRef CAS.
- Review: A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864 Search PubMed.
- S. Heinen and L. Walden, Angew. Chem., Int. Ed., 2000, 39, 806 CrossRef CAS.
Footnotes |
| † Molecular Saddles Part 8: For part 7 of this series see: C. A. Christensen, A. S. Batsanov, M. R. Bryce and J. A. K. Howard, J. Org. Chem., 2001, 66, 3313. |
| ‡ Electronic supplementary information (ESI) available: molecular models of 10 and 1012+ viewed along the plane of the benzene core (Fig. S1) and 18 and 1824+ viewed perpendicular to the plane of the benzene core (Fig. S2). See http://www.rsc.org/suppdata/jm/b1/b106010b/ |
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.