Selective synthesis of fluorinated ethers by addition reaction of alcohols to fluorinated olefins in water
Junji Murataa, Masanori Tamurab and Akira Sekiyab
aResearch Institute of Innovative Technology for the Earth (RITE), c/o AIST Tsukuba Central 5-2, 1-1-1 Higashi, Tsukuba, Ibaraki, 305-8565, Japan. E-mail: jmurata@mx6.nisiq.net
bNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5-2, 1-1-1 Higashi, Tsukuba, Ibaraki, 305-8565, Japan
First published on 1st February 2002
Abstract
The green process for the preparation of fluorinated ether by the addition reaction of 2,2,2-trifluoroethanol to fluorinated olefins was examined. The selective synthesis of fluorinated ether was achieved by increasing the amount of the proton source in the reaction. The reaction with water as the proton source was a particularly environmentally friendly process, because a highly selective synthesis was achieved by means of a completely organic solvent-free procedure. Also, it allowed for a simple work-up, and a scaling up was easily performed.
Green ContextHydrogen-containing fluorinated ethers may contribute towards the development of replacements for CFCs. Some of these molecules look to be well-suited to such a role, and new cleaner methods for their synthesis are required. This paper describes an aqueous route to the products, which involves a selective high-yielding addition reaction carried out in water, with the product being isolated by direct distillation. Such an approach involves mild conditions and little waste.DJM |
Introduction
We have developed hydrogen-containing fluorinated ethers (HFEs) as alternatives to chlorofluorocarbons (CFCs) and hydrochlorofluorocarbons (HCFCs).1 HFEs have almost zero ozone depleting potential (ODP) due to the absence of a chlorine atom in the molecules.2 As well, some HFEs have low global warming potential, and are expected to act as environmentally friendly alternatives.There are many methods of preparing fluorinated ethers. The typical methods are as follows: (i) fluorination of the ether compound by F2 gas,3,4 (ii) fluorination of the ether compound by metal fluoride,5 (iii) electro-chemical fluorination of the ether compound,6 (iv) addition reaction of alcohol to fluorinated olefin under basic conditions,7 and (v) nucleophilic reaction of fluorinated alkoxide.8–12 Of these reactions, the addition reaction of alcohols to fluorinated olefins is the most convenient method to prepare HFEs from industrially available alcohols and fluorinated olefins.
Many addition reactions of alcohols to fluorinated olefins have previously been reported.13–21 In these reports, the reactions were carried out with or without the organic solvent, and unsaturated ether might be formed besides fluorinated ether. In some reports, the reaction mixture was treated with bromine, and then distilled to remove the unsaturated ether.13,17 The removal of the unsaturated ether is easily performed, but the waste of the brominated compound is unavoidable and the production of the unsaturated ether lowers the yield of the saturated ether. In this paper, we investigated the addition reaction of 2,2,2-trifluoroethanol to fluorinated olefins, and achieved the selective synthesis of saturated ether. In addition, the mechanism for the production of unsaturated ether is discussed.
Results and discussion
Addition reactions of 2,2,2-trifluoroethanol to fluorinated olefins
The addition reactions of 2,2,2-trifluoroethanol 1 to fluorinated olefins 2 were carried out (Scheme 1, Table 1). In these reactions, potassium hydroxide was employed as a basic catalyst. | ||
| Scheme 1 Addition reactions of 2,2,2-trifluoroethanol 1 to fluorinated olefins. | ||
| Yield (%)b | ||||||
|---|---|---|---|---|---|---|
| Entry | 2 | Solvent | Temp./°C | Time/h | 3 | 4 |
| a Reaction conditions: 1 (1.0 mmol), 2 (7.5 mmol), solvent (none or 1.0 ml), KOH (1.0 mmol).b Yields were determined by NMR and based on 1.c 1 (3.0 mmol), 2 (1.0 mmol). Yield was based on 2.d H2O (0.1 ml).e CH3COOCH2CF3 was obtained in 6% yield..14f KOH (0.2 mmol).g Mixture of cis and trans isomers. | ||||||
| 1 | CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2 (2a) CF2 (2a) | None | 70 | 24 | 90 | 0 |
| 2 | CF2CF2 (2a) | 1,4-Dioxane | 70 | 0.5 | 99 | 0 |
| 3 | CF2CHF (2b) | None | 70 | 24 | 75 | 9g |
| 4 | CF2CHF (2b) | 1,4-Dioxane | 70 | 0.5 | 58 | 42g |
| 5c | CF2CHF (2b) | 1 | 70 | 24 | 81 | 8 |
| 6d | CF2CHF (2b) | H2O | 70 | 24 | 85 | 1 |
| 7e | CF2CH2 (2c) | None | 70 | 24 | 46 | 44 |
| 8 | CF2CH2 (2c) | 1,4-Dioxane | 70 | 0.5 | 16 | 83 |
| 9c | CF2CH2 (2c) | 1 | 70 | 24 | 22 | 13 |
| 10d | CF2CH2 (2c) | H2O | 70 | 24 | 81 | 3 |
| 11 | CF2CFCF3 (2d) | None | 25 | 24 | 70 | 18g |
| 12 | CF2CFCF3 (2d) | 1,4-Dioxane | 25 | 0.5 | 69 | 29g |
| 13c | CF2CFCF3 (2d) | 1 | 25 | 24 | 84 | 0 |
| 14d | CF2CFCF3 (2d) | H2O | 25 | 24 | 83 | 3 |
| 15df | CF2CFCF3 (2d) | H2O | 25 | 24 | 92 | 1 |
At first, the ratio of saturated ether 3 and unsaturated ether 4 was estimated in the addition reactions without the solvent or in 1,4-dioxane. In the addition reactions with tetrafluoroethylene 2a, 3a was obtained as the only product (Table 1, entries 1 and 2). In the reactions with trifluoroethylene 2b, vinylidene fluoride 2c or hexafluoropropene 2d, not only 3 but also 4 was obtained (Table 1, entries 3, 4, 7, 8, 11 and 12). In the reactions to 2d, some components with high molecular weight were detected by GC-MS. It was considered that these compounds were formed by the addition reaction of 1 to 4d,22 and the generation of these compounds lowered the yield of 3d. In the reaction to 2b or 2d using an excess amount of 1 as the solvent, the yield and the selectivity of 3 were higher than that without the solvent or in 1,4-dioxane (Table 1, entries 5 and 13). Under the same conditions, the reaction to 2c did not proceed efficiently, because a high pressure of 2c was necessary (Table 1, entry 9). In the reaction to 2b, 2c or 2d, the reactions were carried out in water. In any of these cases, the yield and the selectivity of 3 were quite high (Table 1, entries 6, 10 and 14). By using a smaller amount of potassium hydroxide, the yield and the selectivity of 3d were further improved (Table 1, entry 15). The reactions of other fluorinated alcohols to hexafluoropropene were examined briefly. For CF3CF2CH2OH or (CF3)2CHOH, high yield and selectivity were also obtained.
In the reactions to any fluorinated olefins, the use of an excess amount of alcohol or water as the solvent was effective for the selective synthesis of the saturated ether. However, when using an excess amount of the alcohol as the solvent, only some fluorinated olefins reacted efficiently, and it was necessary to isolate the product from the solvent. The reaction in water is a completely organic solvent-free procedure, and a simple work-up can be performed. The fluorinated ether can be extracted easily after the reaction, because the layer of fluorinated ether and the aqueous layer can be separated.
Mechanism for production of unsaturated ether
To authenticate the solvent effect, the mechanism for the production of unsaturated ether 4 was examined. There are two plausible mechanisms for the production of 4. One is the dehydrofluorination of 3 (Scheme 2, path 1), while the other is the elimination of fluoride anion from the carbanion intermediate (Scheme 2, path 2). | ||
| Scheme 2 Mechanism for production of unsaturated ether. | ||
To clarify the contribution of path 1, the stabilities of saturated ethers under the basic conditions were observed (Table 2). The conditions were as follows: a mixture of 3 (1.0 mmol), 1 (1.0 mmol) and potassium hydroxide (1.0 mmol) was stirred. 3b was recovered quantitatively and no unsaturated ether was detected (Table 2, entries 1 and 2). 3c was recovered in 95% yield, but no 4c was detected (Table 2, entry 3). It was presumed that the 5% loss of 3c was due to the hydrolysis of CH3COOCH2CF3 formed by the decomposition of 3c. For 3d in 1,4-dioxane, 3d was recovered with only 72% yield, and the reaction mixture contained a small amount of 4d and some components with high molecular weight (Table 2, entry 6). However, 3d was recovered quantitatively in the case of no solvent or in water (Table 2, entries 5 and 7).
| Entry | CF3CH2OCF2CHXY (3) | Solvent | Time/h | Temp./°C | Recovery of 3 (%) |
|---|---|---|---|---|---|
| 1 | CF3CH2OCF2CH2F (3b) | None | 24 | 70 | 100 |
| 2 | CF3CH2OCF2CH2F (3b) | 1,4-Dioxane | 0.5 | 70 | 100 |
| 3 | CF3CH2OCF2CH3 (3c) | None | 24 | 70 | 95 |
| 4 | CF3CH2OCF2CH3 (3c) | 1,4-Dioxane | 0.5 | 70 | 100 |
| 5 | CF3CH2OCF2CHFCF3 (3d) | None | 24 | 25 | 100 |
| 6 | CF3CH2OCF2CHFCF3 (3d) | 1,4-Dioxane | 0.5 | 25 | 72 |
| 7 | CF3CH2OCF2CHFCF3 (3d) | H2O | 24 | 25 | 100 |
From these results regarding the stabilities of the saturated ethers, only in the addition reaction of 1 to 2d in 1,4-dioxane, would unsaturated ether be formed via both paths 1 and 2, but in the other reactions, path 2 is the exclusive mechanism. Thus, path 2 must be effectively avoided for the selective synthesis of saturated ethers; this was achieved by increasing the amount of the proton source such as water or alcohol in the reaction.
Scaling up
It was confirmed that the addition reaction in water was remarkably effective for the selective synthesis of saturated ethers. In industrial production, this type of reaction could be carried out by feeding the gas of fluorinated olefin to the mixture of the alcohol, the basic catalyst and the solvent during the reaction, because this procedure does not require a large apparatus. The addition reaction of 1 to 2d was then carried out by means of this procedure (Scheme 3, Table 3). To quantify the effect of water in this procedure, reactions without the solvent or in 1,4-dioxane were also carried out. In the latter cases, 3d was obtained in only a fair yield, and 4d and some components with high molecular weight were also obtained ( Table 3, entries 1 and 2). In using water as the solvent, 3d was obtained in an almost quantitative yield ( Table 3, entries 3 and 4). Although water works as a poor solvent and prevents fluorinated olefin from being dissolved to alcohol, the reaction rate could be accelerated by increasing the ratio of 1 to water.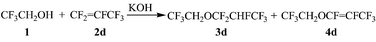 | ||
| Scheme 3 Scaling up addition reactions of 2,2,2-trifluoroethanol to hexafluoropropene. | ||
Experimental
General
All organic reagents and solvents were purified prior to use. Fluorinated olefins were used without purification. KOH was based on 86% purity. 1H and 19F NMR were measured using a JNM-LA300 instrument (JEOL, 300 MHz) and employing TMS and CFCl3 as an internal standard and CDCl3 as a solvent. MS spectra were measured using the Hewlett-Packard 5790 series system equipped with a jet separator for the 5890A GC. HRMS spectra were measured using Hitachi M-80B.Typical procedure for the addition reaction
2,2,2-Trifluoroethanol (1.0 mmol), potassium hydroxide (0.1 mmol) and the solvent were placed in a stainless-steel reactor equipped with a stop valve (volume: 10 ml). After cooling the reactor with liquid N2, fluorinated olefin (7.5 mmol) was introduced to the reactor with a vacuum line and stirred. Products were transferred from the reaction mixture and fractionated through traps at −60, −114 and −196 °C with a vacuum line. The saturated ether and the unsaturated ether were trapped at −114 °C. The structures of the products were determined by 1H, 19F and MS spectra. The ratio of the products was determined by 1H and 19F NMR spectra.The synthesis of 3a–d,184a,234b24 and 4c25 have previously been reported. However, the spectroscopic data for 4c have not been reported, while 4d is a new compound.
Preparation of unsaturated ethers
Sodium 2,2,2-trifluoroethoxide (1.0 mmol) and dried diglyme (1.0 ml) were placed in a stainless-steel rector equipped with a stop valve (volume: 10 ml). After cooling with liquid N2, fluorinated olefin (2.0 mmol) was introduced to the reactor with a vacuum line. The reactor was warmed to ambient temperature and stirred for 24 h. Products were transferred from the reaction mixture and fractionated through traps at −60, −114 and −196 °C with a vacuum line. The unsaturated ether was trapped at −114 °C. The structures of the products were determined by 1H, 19F NMR and MS spectra.1-Fluoro-1-(2,2,2-trifluoroethoxy)ethene 4c
This compound was prepared from vinylidene fluoride: yield 81%.δH (CDCl3, 300 MHz, Me4Si) 3.55 (d of d, 1H, J = 4.7, 39.6 Hz), 3.83 (d of d, 1H, J= 4.7, 5.5 Hz), 4.19 (q, 2H, J = 7.9 Hz), δF(CDCl3, 282 MHz, CFCl3) −74.6 (d of t, 3F, J = 1.2, 7.9 Hz), −84.1 (d of d of q, 1F, J = 1.2, 5.5, 39.6 Hz). MS: m/z, 144 (M+), 83 (CF3CH2+), 45 (CH2CF+), 42, 33.
1,1,1,2,3-pentafluoro-3-(2,2,2-trifluoroethoxy)-2-propene 4d
This compound was prepared from hexafluoropropene: yield 70% (cis/trans = 33/67).Typical procedure for evaluation of stability of saturated ether
Saturated ether (1.0 mmol), 2,2,2-trifluoroethanol (1.0 mmol), potassium hydroxide (0.1 mmol) and the solvent were placed in a stainless-steel rector equipped with a stop valve (volume: 10 ml) and stirred. Products were transferred from the reaction mixture and fractionated through traps at −60, −114 and −196 °C with a vacuum line. The saturated ether was trapped at −114 °C.Typical procedure for scaling up reaction
2,2,2-Trifluoroethanol (0.1 mol), potassium hydroxide (0.1 mol) and the solvent (50 ml) were placed in a stainless-steel reactor (volume: 250 ml). After cooling the reactor with liquid N2, the reactor was warmed up to ambient temperature. Hexafluoropropene was introduced to the reactor and fed continuously during the reaction. The pressure of the hexafluoropropene was kept constant, and the temperature was controlled by cooling the reactor. After stopping the consumption of hexafluoropropene, the organic layer was extracted. The ratio of the products was determined by 1H and 19F NMR spectra.Acknowledgements
We thank the New Energy and Industrial Technology Development Organization (NEDO) for its financial support.References
- A. Sekiya and S. Misaki, J. Fluorine Chem., 2000, 101, 215 CrossRef CAS.
- A. R. Ravishankara, A. A. Turnipseed, N. R. Jensen, S. Barone, M. Mills, C. J. Howard and S. Solomon, Science, 1994, 263, 71 CAS.
- A. Sekiya and K. Ueda, Chem. Lett., 1990, 609 CAS.
- H. S. Huang, D. F. Persico and R. J. Lagow, J. Org. Chem., 1988, 53, 78 CrossRef CAS.
- M. Brandwood, P. L. Coe, C. S. Ely and J. C. Tatlow, J. Fluorine Chem., 1975, 5, 521 CrossRef CAS.
- T. Abe, E. Hayashi, H. Baba, K. Kodaira and S. Nagase, J. Fluorine Chem., 1980, 15, 353 CrossRef CAS.
- R. D. Chambers and R. H. Mobbs, Adv. Fluorine Chem., 1965, 4, 50 Search PubMed.
- J. A. Young and P. Tarrant, J. Am. Chem. Soc., 1950, 72, 1860 CrossRef CAS.
- A. L. Henne and M. A. Smook, J. Am. Chem. Soc., 1950, 72, 4378 CrossRef CAS.
- S. Langner and P. Rollinson, Ger. Pat., 814
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 493, 1959.
493, 1959. - O. Scherer, H. Millauer and B. Wojetch, Ger. Pat., 1283820, 1968.
- O. Scherer and H. Millauer, Ger. Pat.,
1294949, 1969.
- I. L. Knunyants, A. I. Shchekotikhin and A. V. Fokin, Izv. Akad. Nauk. SSSR, Ser. Khim., 1953, 282 Search PubMed.
- J. D. Park, W. M. Sweeney, S. L. Hopwood Jr. and J. R. Lacher, J. Am.. Chem. Soc., 1956, 78, 1685 CrossRef CAS.
- J. L. Rendall and W. H. Oearlson, US Pat., 2862024, 1958.
- A. V. Fokin, V. A. Komarov, A. F. Kolomiets, A. I. Rapkin, O. V. Verenikin and T. M. Potarina, Izv. Akad. Nauk. SSSR, Ser Khim., 1977, 9, 2141 Search PubMed.
- A. V. Gubanov, V. A. Tumanova and I. M. Dolgopolskii, Zh. Obshch. Khim., 1965, 35, 399 Search PubMed.
- R. E. A. Dear and E. E. Gilbert, J. Chem. Eng. Data, 1969, 14, 493 CrossRef CAS.
- R. E. A. Dear and E. E. Gilbert, US Pat., 3557294, 1971.
- S. Tono, A. Nakahara and Y. Izeki, , Jpn. Pat., 9263559, 1997.
- J. D. Park, W. M. Sweeney, S. L. Hopwood Jr. and J. R. Lacher, J. Am. Chem. Soc., 1956, 78, 1685 CrossRef CAS.
- The highest m/z of some components with high molecular weight were 311 or 310 by GC-MS. It was suspected that the peak (m/z: 311) was derived from the fragment ion [M − F]˙+ peak of (CF3CH2O)2CFCHFCF3 or CF3CH2OCHFCF(OCH2CF3)CF3, and the peak (m/z: 310) was derived from the molecular ion peak of (CF3CH2O)2CCFCF3 or CF3CH2OCFC(OCH2CF3)CF3.
- M. J. Pellerite, J. Fluorine Chem., 1990, 49, 43 CrossRef CAS.
- M. H. Hung and S. Rozen, US Pat., 5350497, 1994.
- L. S. Croix and A. J. Buselli, , US Pat., 2799712, 1957.
| This journal is © The Royal Society of Chemistry 2002 |