Absolute photolysis cross-sections for NaHCO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) , NaOH, NaO, NaO2 and NaO3: implications for sodium chemistry in the upper mesosphere
, NaOH, NaO, NaO2 and NaO3: implications for sodium chemistry in the upper mesosphere
Received
6th August 2001
, Accepted 5th October 2001
First published on 18th December 2001
Abstract
The compounds NaHCO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) , NaOH, NaO and NaO2 are formed from meteor-ablated Na in the upper mesosphere. This paper reports the absolute photolysis cross-sections of these species at selected wavelengths between 193 and 423 nm, measured by laser photofragment spectroscopy. The sodium species were produced in both a slow flow reactor and a fast flow tube, by the addition of appropriate reagents to a flow of atomic Na from a heat-pipe oven. The pulsed photolysis source was either a Raman-shifted excimer laser or a Nd:YAG laser, and the resulting Na atoms were probed by laser induced fluorescence at 589.0 nm (Na(3 2P3/2–2S1/2)). Measurements are reported at 200 and 300 K. A limited set of cross-sections for NaO3 was also obtained at 300 K. In the case of NaHCO3, NaOH and NaO, the onset of photolysis is close to the thermodynamic threshold calculated
from quantum theory. Calculations using the CI-singles method predict excited states whose relative energies are in sensible accord with the bands in the experimental spectra, but which are too high in energy with respect to their respective ground states. Photodissociation coefficients are then derived which are appropriate for modelling sodium chemistry in the upper mesosphere. Photolysis of the major sodium reservoir species, NaHCO3, is shown to be an important route for recycling this compound, and is also predicted to produce the HCO3 radical, which should be stable under mesospheric conditions.
, NaOH, NaO and NaO2 are formed from meteor-ablated Na in the upper mesosphere. This paper reports the absolute photolysis cross-sections of these species at selected wavelengths between 193 and 423 nm, measured by laser photofragment spectroscopy. The sodium species were produced in both a slow flow reactor and a fast flow tube, by the addition of appropriate reagents to a flow of atomic Na from a heat-pipe oven. The pulsed photolysis source was either a Raman-shifted excimer laser or a Nd:YAG laser, and the resulting Na atoms were probed by laser induced fluorescence at 589.0 nm (Na(3 2P3/2–2S1/2)). Measurements are reported at 200 and 300 K. A limited set of cross-sections for NaO3 was also obtained at 300 K. In the case of NaHCO3, NaOH and NaO, the onset of photolysis is close to the thermodynamic threshold calculated
from quantum theory. Calculations using the CI-singles method predict excited states whose relative energies are in sensible accord with the bands in the experimental spectra, but which are too high in energy with respect to their respective ground states. Photodissociation coefficients are then derived which are appropriate for modelling sodium chemistry in the upper mesosphere. Photolysis of the major sodium reservoir species, NaHCO3, is shown to be an important route for recycling this compound, and is also predicted to produce the HCO3 radical, which should be stable under mesospheric conditions.
Introduction
The layer of Na atoms that occurs globally between 80 and 105 km is formed by the ablation of sodium from the approximately 120 tons of interplanetary dust that enters the atmosphere each day.1,2Fig. 1 is a schematic diagram of the chemistry that is believed to control the layer.3 Ion–molecule chemistry is important above the peak of the layer at about 90 km, whereas below this altitude Na is oxidised to form a number of neutral species including NaO, NaO2, NaOH and NaHCO3. A recent laboratory study of the reaction between NaHCO3 and H has confirmed that the bicarbonate is the most stable reservoir for sodium immediately beneath the layer.4 Recent models of the Na layer, using rate coefficients that have all now been measured individually under conditions appropriate to the mesosphere, produce excellent
agreement with the observed seasonal and latitudinal variations of the layer.3,5
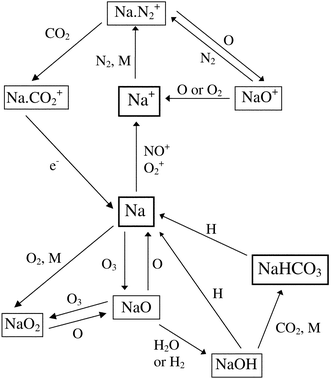 |
| | Fig. 1 The gas-phase chemistry of sodium in the upper atmosphere (adapted from ref. 3). Minor constituents such as NaCO3 and NaO3 have been omitted for clarity. | |
However, these observations were almost exclusively of the night-time Na layer. Recent advances in lidar technology permit fully diurnal measurements of the layer to be made on a routine basis, yielding both Na density and temperature (from the broadening of the hyperfine structure of the D2 line at 589.0 nm).6 In order to explain the complex diurnal variability of the Na density profile, several coupled factors need to be taken into account. First, there is the effect of tidal dynamics on the layer. Indeed, the amplitudes and phases of the diurnal and semi-diurnal tides have been obtained from analysis of the Na temperature record.6 Second, the odd oxygen and hydrogen chemistry, which controls the underside of the Na layer (Fig. 1), exhibits a large diurnal variation below 82 km where the concentrations of O and H vary by orders of magnitude. Finally, the
sodium compounds themselves may photolyse significantly during daytime, contributing a further diurnal oscillation. This is the subject of the present study.
In this paper we will describe measurements of the absolute photolysis cross-sections (to yield atomic Na) of the neutral Na-containing molecules in Fig. 1, using a laser photofragmentation method. Ab initio quantum calculations will then be employed to interpret the results, before calculating photodissociation coefficients suitable for use in mesospheric models. These sodium compounds have very large dipole moments and therefore readily cluster with water, particularly at the low temperatures of the upper mesosphere during summer.7 The effect of this on the photolysis cross-sections will also be examined.
Experimental measurements
The absolute photolysis cross-sections of NaHCO3, NaOH, NaO, NaO2 and NaO3 were determined by the technique of pulsed laser photolysis followed by laser-induced fluorescence (LIF) of the resulting Na atoms. This photofragmentation technique has been used previously by Silver et al.8 and Rajasekhar et al.9 to study the photolysis of NaCl and NaO2, respectively. The sodium compound NaX (where X=HCO3, OH, O, O2 or O3) is produced in situ by essentially complete conversion of a flow of Na atoms entering the reactor from an oven, following the addition of appropriate reagents in large excess:| | 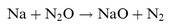 | (R1) |
| | 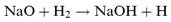 | (R2) |
| | 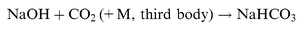 | (R3) |
| | 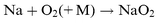 | (R4) |
| | 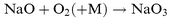 | (R5) |
If the resulting flow of NaX is subjected to a laser pulse of fluence I0(λ) at wavelength λ, then in the limit of low light absorption the concentration of nascent Na atoms created by the pulse is given by| | 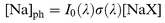 | (I) |
where σ(λ) is the absolute photolysis cross-section at λ to yield Na+X. Note that we assume here that the photolysis quantum yield following absorption of a photon is unity. This will be justified below. The Na atoms will give rise to a measured LIF signal, Ifp, when the probe laser is triggered immediately afterwards. In the absence of reagents, the LIF signal (If0)
will be proportional to the total Na atom concentration. This is equivalent to [NaX] when reagents are added, after making a small correction for differences in the rates of diffusion of Na and NaX (see below). Since Ifp and If0 are measured with the same geometrical light collection factor and detection sensitivity, then| | 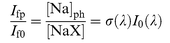 | (II) |
Thus, σ(λ) can be determined absolutely by measuring the ratio Ifp/If0, and monitoring I0(λ).
Two experimental systems were used in this study. The first, illustrated schematically in Fig. 2, employed a stainless steel slow flow reactor that has been described in detail elsewhere.9 The central chamber of this reactor is at the intersection of two sets of side-arms that cross orthogonally. One set of arms provides the optical coupling of the lasers to the central chamber, as well as the means by which the flows of the reagents enter the chamber. A third side-arm was independently heated (520–555 K) to provide a source of Na atoms. A spiral of stainless steel mesh (gauge 150) was inserted into this heat pipe, along with solid Na spheres. Upon heating the mesh became wetted with molten Na, thereby greatly increasing the surface area exposed to the flow of N2 gas that entrained the Na vapour into the central chamber. This feature, together with accurate control of the heat pipe temperature (±1
K), ensured a stable flow of Na atoms during the experiments.
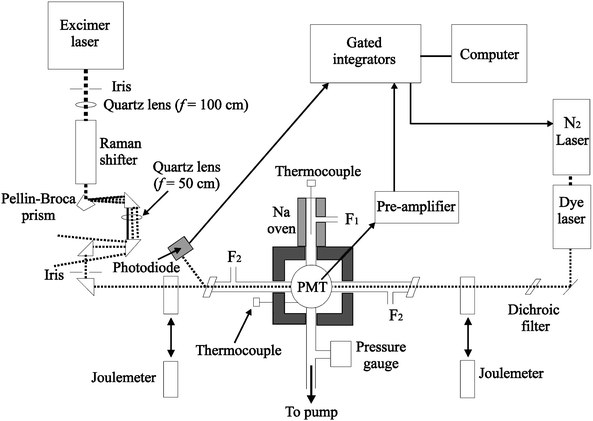 |
| | Fig. 2 Schematic diagram of the slow flow reactor used to measure the absolute photolysis cross-sections of Na-containing molecules by photofragment spectroscopy: F1, flow of N2 through the sodium oven; F2, flow of reagents into the central chamber; PMT, photomultiplier tube. | |
The central chamber is enclosed in an insulated compartment that can be filled with dry ice to cool it to about 200 K. The temperature of the gas flow in the chamber is monitored by a thermocouple permanently inserted about 1 cm below the path of the laser beams. Once in the chamber, the atomic Na was mixed with the reagent flows at a total pressure of about 3 Torr. The synthesis of NaHCO3via reactions (R1)–(R3) required some care. On the one hand, sufficient CO2 had to be added to ensure that all the NaOH produced by (R2) was converted to NaHCO3; on the other, competition for NaO from the reaction
| | 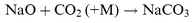 | (R6) |
had to be minimised. A full kinetic model
4 was employed to optimise the reagent mixing ratios: at 300 K the reagents N
2O/H
2/CO
2/N
2
were typically added in the ratio 5 : 20 : 0.3 : 74.7 (at 200 K, [N
2O] was increased because reaction (
1) has a significant activation energy
10).
Photolysis was carried out over a range of wavelengths from 193 to 423 nm, using a Raman-shifted excimer laser (Questek Model 2010, with unstable resonator optics) operating at 193 nm (ArF) or 248 nm (KrF). The Raman shifter (Lambda Physik, model RS75) was filled with H2
(Air Products, 99.9999%) or D2
(Messer, 99.7%) to between 2.2 and 10 bar. The output beams were separated spatially using a Pellin–Broca prism. The fluences of the Raman-shifted beams were measured before and after the reactor using a Molectron J-25 Joulemeter (5 mm diameter circular element, calibrated to a recent factory reference) and a quartz beamsplitter. The fluence in the central chamber was then computed as the geometric mean of the two measurements. The Raman-shifted wavelengths and typical laser fluences in the reactor are listed in Table 1.
Table 1 Typical Raman-shifted excimer laser fluences in the reactora
| |
H2 in Raman shifter |
D2 in Raman shifter |
| |
λ/nm |
Fluence/mJ cm−2 |
λ/nm |
Fluence/mJ cm−2 |
| |
|
An=Anti-Stokes, shifted n times, Sn=Stokes, P=primary.
|
| KrF |
|
|
|
|
| A2 |
205.9 |
0.04 |
216.3 |
0.3 |
| A1 |
225.2 |
2 |
231.2 |
2 |
| P |
248.4 |
20 |
248.4 |
40 |
| S1 |
277.0 |
8 |
268.3 |
7.5 |
| S2 |
313.0 |
6 |
291.8 |
5 |
| S3 |
359.8 |
3.5 |
319.6 |
3.5 |
| S4 |
423.0 |
0.7 |
353.4 |
1.5 |
| S5 |
513.2 |
0.05 |
395.2 |
0.3 |
| S6 |
652.3 |
0.005 |
448.2 |
Unstable |
| ArF |
|
|
|
|
| P |
193.4 |
5 |
193.4 |
3 |
| S1 |
210.3 |
3 |
205.3 |
1.2 |
| S2 |
230.4 |
2.5 |
218.7 |
0.9 |
| S3 |
254.8 |
1.5 |
234.0 |
0.6 |
| S4 |
285.0 |
0.3 |
251.6 |
0.3 |
The relative atomic Na concentration was measured by pumping one of the Na D-lines at 589.0 nm [Na(3 2P3/2–3 2S1/2)] using a N2-pumped dye laser, and observing non-resonant LIF at 589.6 nm through a narrowband interference filter (Janos Corp., FWHM=0.2 nm). A vertical side arm on the chamber provided the optical coupling to the photomultiplier tube (Thorn EMI Gencom, model 9816QB). The signal was captured by a gated integrator after pre-amplification (Stanford Research Systems, model SR445, 300 MHz). The linearity of the measured LIF signal with the Na atom density was regularly checked.
During photolysis experiments, the dye laser triggered 8.7 µs after the photolysis pulse. During this period an insignificant fraction of the nascent Na atoms would have reacted with the oxidant (N2O or O2) in the chamber. When the Na concentration in the absence of reagents was being measured (i.e.If0), the total flow and pressure in the central chamber were maintained by increasing the N2 flow through the side-arms. In order to make a measurement of σ(λ), Ifp and If0 were obtained by averaging a minimum of 200 data points in each case. A background (zero) signal was also obtained by measuring the LIF signal with the reagents flowing through the reactor but the photolysis laser off.
A limited number of experiments were performed using a second experimental system. This was a fast flow tube, described recently by Cox et al.,4 which operated at a pressure of 1.5 Torr with flow velocities of 12–24 m s−1. Photolysis cross-sections were determined using the same photolysis/LIF technique. Na vapour was produced in an oven at the upstream end of the tube, entrained in N2 and then mixed in the flow tube with reagents that were added sequentially to produce the same compounds as before. Photolysis cross-sections were determined using a frequency tripled or quadrupled Nd:YAG laser (Continuum Minilite). The beam energy was determined with a calibrated Molectron J-50 Joulemeter, and the beam area measured with photosensitive paper. This yielded fluences of 170 mJ cm−2 at 354.6 nm and 110 mJ cm−2 at 265.9 nm.
The cross-section measurements with both experimental systems were performed with a total Na concentration less than 1×109 molecule cm−3 in the photolysis region. This is important in order to avoid the clustering of Na compounds to form dimers and higher polymers. Polymerisation should be a fast process because of the strong intermolecular forces resulting from the large dipole moments of these species (see below). The absolute Na concentration was determined in a separate experiment where it was measured in the absence of reactants by the resonance absorption of the radiation from a Na hollow cathode lamp directed transversely to the flow. An absorption cross section of 1.85×10−11 cm2 for the [Na(3 2PJ–3 2S1/2)] transition was assumed.11
Materials
N2
(BOC, 99.9999% pure), N2O (Air Products, 99.99% pure), CO2
(Air Products, 99.995% pure), H2
(Air Products, 99.9999% pure) and O2
(Air Products, 99.995% pure) were used without further purification. Sodium metal spheres (3–8 mm, Aldrich Chem. Co., Inc.) were cleaned with solvent before insertion into the heat-pipe ovens.
Results
The photolysis cross-sections, σ(λ), were determined using eqn. (II). Measurements of the NaHCO3, NaOH, NaO and NaO2 cross-sections were made at both 300 and 200 K, since the lower temperature is representative of the upper mesosphere. The photolysis of NaO3, which is a less important mesospheric species,3 was only studied at 300 K. Diffusion corrections to σ(λ) were made in the following way. For the slow flow reactor (Fig. 1), the flow of Na atoms from the heat pipe into the central chamber is balanced by diffusion to the chamber walls of Na (in the absence of reagents) or of NaX. As we have shown previously for this reactor,12 the steady-state concentration of Na is given by FNa/kdiff(Na),
where FNa is the flow rate of Na into the chamber from the heat pipe, and kdiff(Na) is the first-order loss of Na to the walls of the chamber, which closely approximates to a cylinder of radius r=3 cm and length l=6 cm. kdiff(Na) is given by| | 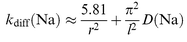 | (III) |
where D(Na) is the diffusion coefficient for Na in N2, at the pressure and temperature of the central chamber. When reagents are added to the same flow of Na from the heat pipe, the steady-state concentration of NaX is by analogy FNa/kdiff(NaX). Hence, the cross-section σ(λ) needs to be corrected by the factor kdiff(Na)/kdiff(NaX)≈D(Na)/D(NaX).
The diffusion coefficients used to make this correction are listed in Table 2. When correcting the 200 K data, it was assumed that the diffusion coefficients of Na and NaX had the same temperature dependences.
Table 2 Diffusion coefficients and thermodynamic data
| Species |
Diffusion coefficient at 300 K/cm2 s−1 Torr |
Thermo-dynamic limit λ/nm |
Lowest vibrational frequency/cm−1 |
Ratio of v′′=1 populations 300 to 200 K |
| |
|
Ref. 4.
Measured relative to D(Na) in the fast flow tube.
Estimated from long-range capture theory (see ref. 14).
Ref. 4 and present calculations on HCO3.
Ref. 17.
Ref. 18.
Ref. 22.
Ref. 23.
|
| NaHCO3 |
120a |
257d |
118a |
1.33 |
| NaOH |
134b |
368e |
261e |
1.87 |
| NaO2 |
119b |
747f |
333g |
2.22 |
| NaO |
141a |
443e |
492h |
3.26 |
| NaO3 |
110c |
581f |
— |
— |
| Na |
152a |
— |
— |
— |
For the fast flow tube measurements, the diffusion correction was applied by assuming uptake coefficients for Na and NaX of close to unity on the walls so that strong radial concentration gradients would have developed in the tube. The first-order loss to the walls under laminar flow conditions was then described using the approximation of Huggins and Cahn.13 In the case of NaOH and NaO2, a movable injector was used to vary the point in the flow tube at which reagents were added to produce these species from Na. By observing the apparent change in σ(λ) as the injection distance was varied, the diffusional loss to the walls of NaOH and NaO2 was determined relative to that of Na.
The resulting σ(λ) are listed in Tables 3–7 for the five sodium species that were studied. The quoted 1σ errors are computed by combining in quadrature the statistical and systematic errors. The statistical error arises from the standard deviations in the measurement of If0 and Ifp
(eqn. (II)). When using a strong photolysis laser line (e.g. the KrF primary) and if σ(λ)>10−18 cm2, this error was less than 10%. However, if σ(λ) was very small or the laser fluence was weak, then the statistical error could exceed 100%. There are two significant sources of systematic error. First is the uncertainty in the measured laser fluence.
This ranged from 4% for the primary excimer lines to 20% for the highly Raman-shifted beams. The YAG fluences were estimated to have an uncertainty of 13%. The second systematic uncertainty is in the diffusion correction factor. For NaOH and NaO2, the diffusion coefficients relative to D(Na) were measured using the fast flow tube (see above), and D(NaO) was determined in a previous flow tube study,4 so the error in the diffusion correction factors for these species is better than 5%. D(NaHCO3) and D(NaO3) were estimated from long-range capture theory,14 using parameters calculated from quantum theory (see below); the uncertainty in their diffusion correction factors is estimated to be 12%.
Table 3 Photolysis cross-sections for NaHCO3
| Raman linea |
λ/nm |
σ(λ) at 300 K/10−18 cm2 |
σ(λ) at 200K/10−18 cm2 |
| |
|
The nomenclature used to describe the photolysis line: exciplex (ArF or KrF), Raman-shifting gas (H2 or D2), An=anti-Stokes n shifts, Sn=Stokes n shifts, P=primary. Nd:YAG 3ν or 4ν=tripled or quadrupled Nd:YAG laser.
|
| ArF P |
193.4 |
1.13±0.23 |
0.7.35±0.237 |
| KrF H2 A2 |
205.9 |
0.487±0.358 |
|
| ArF H2 S1 |
210.3 |
1.16±0.36 |
0.947±0.387 |
| KrF D2 A2 |
216.3 |
1.20±0.56 |
|
| KrF H2 A1 |
225.2 |
0.773±0.093 |
0.987±0.501 |
| ArF H2 S2+KrF D2 A1 |
230.9 |
0.547±0.179 |
0.578±0.217 |
| KrF P |
248.4 |
0.0401±0.0054 |
0.0355±0.0113 |
| Nd:YAG 4ν |
265.9 |
−0.11±0.15 |
|
| KrF D2 S1 |
268.3 |
0.0095±0.0269 |
|
| KrF H2 S1 |
277.0 |
0.0109±0.0045 |
0.0549±0.0438 |
| Nd:YAG 3ν |
354.6 |
−0.018±0.030 |
|
Table 4 Photolysis cross-sections for NaOH
| Raman linea |
λ/nm |
σ(λ) at 300 K/10−18 cm2 |
σ(λ) at 200K/10−18 cm2 |
| |
|
See Table 3.
|
| ArF P |
193.4 |
5.51±0.43 |
2.92±2.38 |
| ArF D2 S1+KrF H2 A2 |
205.7 |
8.67±2.74 |
|
| KrF H2 A2 |
205.9 |
|
5.63±2.52 |
| ArF H2 S1 |
210.3 |
|
1.85±3.28 |
| KrF D2 A2 |
216.3 |
4.89±2.60 |
|
| ArF D2 S2 |
218.7 |
6.96±2.79 |
3.76±5.12 |
| KrF H2 A1 |
225.2 |
14.3±1.3 |
10.5±1.5 |
| ArF H2 S2+KrF D2 A1 |
230.6 |
18.2±0.18 |
7.66±4.87 |
| ArF D2 S3 |
234.0 |
16.8±5.3 |
|
| KrF P |
248.4 |
1.73±0.13 |
0.580±0.071 |
| ArF H2 S3 |
254.8 |
1.15±0.22 |
|
| Nd:YAG 4ν |
265.9 |
0.48±0.15 |
|
| KrF D2 S1 |
268.3 |
0.418±0.150 |
0.173±0.120 |
| KrF H2 S1 |
277.0 |
0.191±0.027 |
0.0719±0.0791 |
| ArF H2 S4 |
285.0 |
0.071±0.147 |
|
| KrF D2 S2 |
291.8 |
0.438±0.130 |
0.118±0.123 |
| KrF H2 S2 |
313.0 |
5.85±0.47 |
4.72±0.53 |
| KrF D2 S3 |
319.6 |
5.10±1.03 |
3.41±0.78 |
| KrF D2 S4 |
353.4 |
0.539±0.227 |
0.449±0.444 |
| Nd:YAG 3ν |
354.6 |
0.57±0.20 |
|
| KrF H2 S3 |
359.8 |
0.414±0.064 |
0.165±0.067 |
| KrF D2 S5 |
395.2 |
0.512±0.926 |
|
| KrF H2 S4 |
423.0 |
−0.0130±0.0565 |
0.0180±0.0961 |
Table 5 Photolysis cross-sections for NaO
| Raman linea |
λ/nm |
σ(λ) at 300 K/10−18 cm2 |
σ(λ) at 200K/10−18 cm2 |
| |
|
See Table 3.
|
| ArF P |
193.4 |
5.01±2.32 |
|
| KrF H2 A2 |
205.9 |
−0.42±2.46 |
|
| KrF D2 A2 |
216.3 |
14.9±4.2 |
5.15±3.14 |
| KrF H2 A1 |
225.2 |
11.0±2.7 |
|
| KrF D2 A1 |
231.2 |
11.7±2.2 |
3.53±0.75 |
| KrF P |
248.4 |
2.06±0.16 |
0.937±0.157 |
| Nd:YAG 4ν |
265.9 |
1.8±0.28 |
|
| KrF D2 S1 |
268.3 |
1.60±0.26 |
0.246±0.178 |
| KrF H2 S1 |
277.0 |
0.463±0.049 |
|
| KrF D2 S2 |
291.8 |
1.02±0.29 |
0.955±0.532 |
| KrF H2 S2 |
313.0 |
12.5±2.6 |
|
| KrF D2 S3 |
319.6 |
10.7±1.5 |
7.03±1.28 |
| KrF D2 S4 |
353.4 |
2.76±0.67 |
1.19±0.60 |
| Nd:YAG 3ν |
354.6 |
2.8±0.72 |
|
| KrF H2 S3 |
359.8 |
1.74±0.92 |
|
| KrF D2 S5 |
395.2 |
2.44±1.36 |
1.55±1.52 |
| KrF H2 S4 |
423.0 |
0.522±1.03 |
|
Table 6 Photolysis cross-sections for NaO2
| Raman linea |
λ/nm |
σ(λ) at 300 K/10−18 cm2 |
σ(λ) at 200K/10−18 cm2 |
| |
|
See Table 3.
|
| ArF P |
193.4 |
3.83±0.85 |
1.88±0.51 |
| KrF H2 A2 |
205.9 |
1.46±1.84 |
−1.17±2.08 |
| ArF H2 S1 |
210.3 |
5.42±1.25 |
2.65±0.76 |
| KrF D2 A2 |
216.3 |
5.11±2.08 |
1.92±1.22 |
| KrF H2 A1 |
225.2 |
2.35±0.95 |
5.08±0.93 |
| ArF H2 S2+KrF D2 A1 |
230.8 |
9.35±1.35 |
2.84±0.49 |
| KrF P |
248.4 |
4.14±0.88 |
2.86±0.17 |
| ArF H2 S3 |
254.8 |
5.50±1.34 |
2.22±0.58 |
| KrF D2 S1 |
268.3 |
4.58±0.73 |
2.18±0.24 |
| KrF H2 S1 |
277.0 |
1.81±0.42 |
1.63±0.19 |
| ArF H2 S4 |
285.0 |
0.734±0.330 |
0.355±0.189 |
| KrF D2 S2 |
291.8 |
1.68±0.33 |
0.683±0.103 |
| KrF H2 S2 |
313.0 |
1.27±0.41 |
0.955±0.142 |
| KrF D2 S3 |
319.6 |
0.121±0.113 |
0.252±0.096 |
| KrF D2 S4 |
353.4 |
0.162±0.165 |
0.283±0.169 |
| YAG 3ν |
354.6 |
0.35±0.22 |
|
| KrF H2 S3 |
359.8 |
0.610±0.251 |
0.419±0.103 |
| KrF D2 S5 |
395.2 |
1.08±0.94 |
1.16±0.85 |
| KrF H2 S4 |
423.0 |
0.428±0.309 |
0.211±0.105 |
Table 7 Photolysis cross-sections for NaO3
| Raman linea |
λ/nm |
σ(λ) at 300 K/10−18 cm2 |
| |
|
See Table. 3
|
| KrF D2 A2 |
216.3 |
13.1±5.8 |
| KrF D2 A1 |
231.2 |
2.28±0.71 |
| KrF D2 P |
248.4 |
1.19±0.18 |
| KrF D2 S1 |
268.3 |
0.949±0.174 |
| KrF D2 S2 |
291.8 |
1.45±0.25 |
| KrF D2 S3 |
319.6 |
3.45±0.59 |
| KrF D2 S4 |
353.4 |
0.556±0.211 |
| KrF D2 S5 |
395.2 |
0.717±0.565 |
Ab initio
quantum calculations
The experimental measurements were complemented by a series of ab initio quantum calculations using the Gaussian 98 suite of programs.15 The ground-state geometries of NaHCO3, NaOH, NaO and NaO2 and NaO3 were first optimised using Hartree–Fock (HF) theory with the large flexible 6-311+G(2d,p) basis set, which has both polarization and diffuse functions added to the atoms.16 These molecules all have highly ionic ground states with large dipole moments (6.9, 6.6, 8.6, 8.3 and 7.4 D, respectively). We have published the details of their geometries and vibrational frequencies elsewhere.4,17,18 Calculations on the vertical electronic excited state energies and transition oscillator strengths were performed using configuration interaction-singles (CIS) theory.15,16
In order to investigate the photolysis products of NaHCO3, calculations were performed on the HCO3 radical using hybrid density functional/Hartree–Fock B3LYP theory with the 6-311+G(2d,p) basis set. Fig. 3 illustrates the optimised geometry of this species, as well as the saddle-point geometry for dissociation to OH+CO2. Finally, calculations were carried out on the ground and excited states of the NaOH–H2O and NaHCO3–H2O complexes, to examine the effect of water clustering on photolysis. The H2O binding energies were determined using the very accurate complete basis set (CBS-Q) theory,19 and the calculations on the excited states of the clusters were determined at the CIS/6-311+g(2d,p) level. The ground state geometries of these clusters are also illustrated in Fig. 3.
![Molecular geometries predicted by ab initio quantum theory at the B3LYP/6-311+G(2d,p) level: (a) HCO3
(planar); (b) the HCO3 → OH+CO2 saddle point (H–O–C–O[bottom left] dihedral angle = 68°); (c) the NaOH–H2O cluster (H[top right]–O–Na–O dihedral angle = 144°); (d) the NaHCO3–H2O cluster (planar).](/image/article/2002/CP/b107078a/b107078a-f3.gif) |
| | Fig. 3 Molecular geometries predicted by ab initio quantum theory at the B3LYP/6-311+G(2d,p) level: (a) HCO3
(planar); (b) the HCO3→OH+CO2 saddle point (H–O–C–O[bottom left] dihedral angle=68°); (c) the NaOH–H2O cluster (H[top right]–O–Na–O dihedral angle=144°); (d) the NaHCO3–H2O cluster (planar). | |
Discussion
Inspection of Tables 3–6 shows that the cross-sections measured in the flow reactor and the fast flow tube agree very well, even though the Na compounds were prepared somewhat differently in each type of system. Fig. 4 illustrates the photolysis cross-section spectra of NaHCO3, NaOH, NaO2 and NaO. Note that the maximum cross-section for NaHCO3 is a factor of 10 smaller than the maxima in the spectra of the other compounds, and it only begins to absorb quite far into the ultra-violet. Shown on the figure and also listed in Table 2 are the thermodynamic limits to photolysis, calculated from the respective bond energies D0(Na–X). The sources of these bond energies are given in the footnotes to Table 2. For NaHCO3, NaOH and NaO, the thermodynamic
limit is close to the onset of photolysis in each case, implying that the relevant excited states have rather flat potential energy curves i.e. they are essentially covalent and at best weakly bound. This is supported by the CIS quantum calculations, which show that the Mulliken electron population on the Na atom, which is greater than +0.9 in the ground state, is less than +0.1 in these excited states. These calculations indicate that transitions to these covalent states should lead to prompt dissociation to Na atoms.
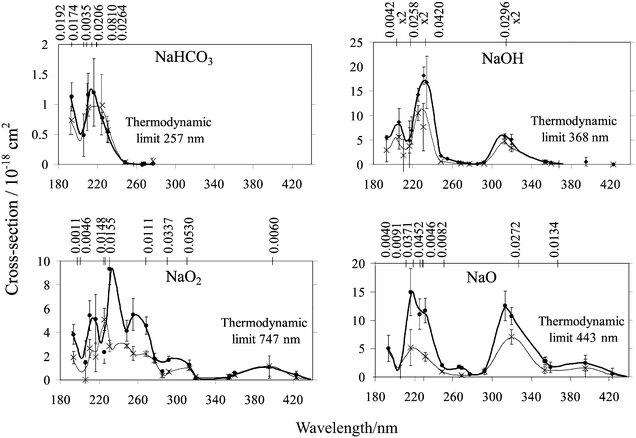 |
| | Fig. 4 Photolysis cross-section spectra of NaHCO3, NaOH, NaO2 and NaO at 200 K (thin line) and 300 K (heavy line). The solid lines drawn through the experimental points are to guide the eye. Error-bars correspond to 1σ uncertainties. The positions and oscillator strengths of the vertical transitions predicted by CIS theory are plotted on the top abscissa of each panel. Note that the excited state energies have been shifted with respect to the ground state (see text). | |
The CIS method, which models excited states as combinations of single substitutions out of the Hartree–Fock ground state, is an approximate treatment for excited states.16 In the present case it systematically over-predicts the vertical excitation energies from the ground state. In order to obtain sensible agreement with the positions of the observed absorption bands, the upper states of each molecule were shifted downwards by the same amount with respect to the ground state. These shifts are listed in Table 8 for the five molecules, and range from 0.6 to 2.7 eV. The (shifted) wavelength positions for transitions where the transition oscillator strength f is greater than 10−3
(and therefore significant), are marked on the top abscissa of each of the panels in Fig. 4 along with their f values. There is reasonable agreement with the
measurements, implying that the relative energies of the covalent upper states are quite well predicted with respect to each other. However, the change from ionic to covalent character between the ground and the upper states is not well described by CIS theory, since the Hartree–Fock orbitals used to describe the excited states are biased to the ground-state configuration.16
Table 8 Energy corrections applied to CIS theory
| Compound |
Energy correction/eV |
|
| |
|
| NaHCO3 |
2.72 |
|
| NaOH |
1.12 |
|
| NaO2 |
0.94 |
|
| NaO |
0.56 |
|
| NaO3 |
0.69 |
|
Fig. 4 shows that some of the bands of NaO and NaO2 exhibit marked positive temperature dependences. A smaller dependence may be present in the case of NaOH, whereas NaHCO3 exhibits no apparent dependence. This is probably explained by vibrational excitation of the ground state molecule leading to better Franck–Condon overlap with its excited states. For example, CIS calculations show that the ground state of NaOH is linear, whereas the covalent excited states are bent: hence, excitation in the degenerate bending modes should increase σ(λ). Table 2 lists the lowest vibrational frequencies of NaHCO3, NaOH, NaO2 and NaO, and the ratios of the Boltzmann population of v′′=1 at 300 K compared to 200 K. This ratio increases markedly from 1.3 for NaHCO3 to 3.3 for NaO, in accord
with the observed variation in temperature dependence shown in Fig. 4.
There is an interesting aspect to the spectrum of NaO in the near ultra-violet. Langhoff et al.20 carried out multi-reference configuration-interaction calculations on the first three states of NaO. This showed that the C 2Π state should be accessible about 25000 cm−1
(≈400 nm) above the X 2Π ground state. Furthermore, because the equilibrium Na–O separation of the C state is about 1.5 Å larger than the X state, the theory predicted that NaO(C–X) absorption would be followed by emission to high vibrational levels of the X state, producing a band system from 500 to 2000 nm and peaking around 1000 nm. However, Pugh et al.21 failed to observe an LIF spectrum for NaO in the 390–430 nm region. Ellis has reported a similar finding.22
The absence of fluorescence is now explained by the present study, which shows that absorption in this region leads to photolysis (Fig. 4). The implication is that NaO(C 2Π) is crossed by a higher-lying 2Π state, causing pre-dissociation to Na+O. Further theoretical work should be carried out on this system. It should also be noted that the reaction between Na and N2O produces NaO in both the X(2Π) and A(2Σ+) states.23,24 There is some evidence that NaO(A 2Σ+) is not quenched very efficiently, and has a radiative lifetime of more than 20 ms.24 Hence, the cross-sections measured in this study may describe a mixture of the X and A states. These are separated by about 2050 cm+1,20,24
corresponding to ≈20 nm in the near-UV, which may explain the rather broad bands in the NaO spectrum (Fig. 4).
The results for NaO2 are in excellent agreement with a previous study using a slow flow reactor.9 The only other work on the absorption spectrum of NaO2 appears to be a matrix isolation study where Na atoms and O2 were co-deposited in argon.25 The resulting superoxide species gave rise to bands with maxima at 232, 250 and 272 nm, which correspond approximately with bands in the 200 K spectrum in Fig. 4.
An absolute absorption spectrum for NaOH in the ultraviolet has been obtained by Rowland and Makide,26 based on spectroscopic data when aqueous NaOH solutions were aspirated into flames between 900 and 2300°C. Fig. 5 is a comparison of the flame absorption cross-section spectrum with the photolysis spectrum from the present study. There is generally good agreement in the absolute magnitudes of the cross-sections, particularly for the strong band around 235 nm and in the absence of absorption between 265 and 290 nm. The discrepancy beyond 310 nm could be caused by hot-band absorption by vibrationally-excited NaOH in the high temperature flames: indeed, Rowland and Makide26 pointed out that the flame absorption spectrum continued beyond the thermodynamic limit set by the D0(Na–OH) bond energy (see below). Another possibility is that the
apparent structure in the flame spectrum beyond 320 nm is actually due to other absorbing species present in the flame that were not accounted for in the analysis. Finally, it should be noted that photolysis at 193 nm of NaOH produces Na(3 2P) which then fluoresces at 589 nm. This is employed as a diagnostic for sodium in combustion systems, where NaOH is the dominant form at temperatures above 1500°C.27 The threshold for Na(2P) production is about 226 nm, which is close to the onset of the third absorption band in the NaOH spectrum in Fig. 4.
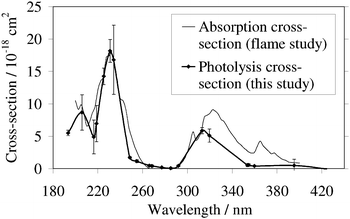 |
| | Fig. 5 The photolysis cross-section of NaOH at 300 K from the present study, compared with an absorption cross-section measured in flames (ref. 25). | |
Atmospheric implications
The photodissociation coefficients for these Na-containing species were computed from the relation:| |  | (IV) |
where Φ(λ) is the solar actinic flux in the upper mesosphere.28 The integration was performed by using a smooth interpolation between the experimental results at discrete wavelengths (see Fig. 4). In the case of NaO2, where photolysis at wavelengths longer than 440 nm is thermodynamically possible, the CIS calculations indicate that there are no low-lying states (Fig. 4). Photolysis at wavelengths shorter than 190 nm will make only a minor contribution to J because the solar flux decreases very rapidly in the vacuum ultra-violet.28 The calculated J values and the lifetimes of the molecules against
photolysis are listed in Table 9. In a previous study of the photolysis of NaO2 where σ(λ) was only measured at three wavelengths,9 the estimate of J(NaO2)=(4.8±2.8) s−1 is about a factor of 2 larger than the result of the present study, although there is agreement within the quoted error.
Table 9 Photolysis J-values and 1/e-lifetimes at 90 km altitude
| Compound |
J(300 K)/ s−1 |
J(200 K)/ s−1 |
τ(300 K)/s |
τ(200 K)/ s |
| |
| NaHCO3 |
1.28±0.47 × 10−4 |
1.27±0.66 × 10−4 |
7800 |
7860 |
| NaOH |
0.0240±0.0077 |
0.019±0.005 |
42 |
54 |
| NaO2 |
0.023±0.013 |
0.019±0.011 |
44 |
53 |
| NaO |
0.093±0.038 |
0.055±0.029 |
11 |
18 |
| NaO3 |
0.026±0.015 |
|
38 |
— |
Fig. 6 compares the calculated J values for the four important Na species with their most rapid chemical loss processes. These involve reaction of atomic H with NaHCO3 and NaOH, atomic O reacting with NaO and NaO2. The conditions are midday in June at 40°N (see the figure caption for further details). In the case of NaO and NaO2, photolysis is not competitive: atomic O is very abundant in the upper mesosphere, and these species react rapidly with O.14,29 In contrast, photolysis of NaHCO3 and NaOH dominates their removal during daytime: atomic H is 2–3 orders of magnitude less abundant than O, and the rates of the reactions of these species with H are quite slow.3,4
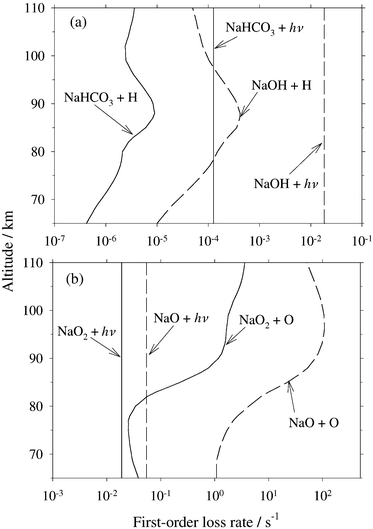 |
| | Fig. 6 Height profiles of the first-order removal rates of (a) NaHCO3 and NaOH by photolysis and reaction with atomic H, and (b) NaO2 and NaO by photolysis and reaction with atomic O. The conditions are midday, June at 40°N. The profiles of O and H are taken from the thermosphere–ionosphere–mesosphere–electrodynamics general circulation model (TIME GCM, ref. 32). | |
In terms of understanding the chemistry of Na in the mesosphere, the photolysis of NaHCO3 is clearly important since this species is believed to be the major gas-phase sodium reservoir below about 85 km.3 With a lifetime against photolysis of only about 2 h, a large diurnal cycle in atomic Na below 85 km would be predicted. Since this is not in fact observed,6 NaHCO3 must be removed from the system, most likely through uptake on meteoric smoke particles.30 We are currently examining this with a new diurnal Na model.
NaHCO3 and NaOH have very large dipole moments (see above), so that they can form stable H2O clusters and this may affect their rates of photolysis. CBS-Q calculations indicate that the binding energies of H2O to NaHCO3 and NaOH are 51.7 and 84.5 kJ mol−1, respectively. As we have shown previously,7 even in the very dry mesosphere with H2O mixing ratios of a few ppm, these (and higher) clusters will dominate once the temperature falls below about 170 K. This situation occurs during mid-summer, particularly at high latitudes. CIS calculations on the excited states of NaHCO3·H2O show that the vertical excitation energies of the first 5 excited states increase by 31–62 kJ mol−1; for NaOH·H2O, the increase is 65–113 kJ mol−1. These energy ranges are consistent with the
respective binding energies of the clusters. This is to be expected, because H2O binds strongly to the ionic ground states of these molecules (Fig. 3), but is weakly bound to the covalent upper states.
To test the effect of cluster formation, we performed a simple experiment on NaOH photolysis in the slow flow reactor. A flow of H2O was added to the central chamber at 200 K, where the equilibrium H2O concentration of 5.3×1013 cm−3 was sufficient to convert the NaOH overwhelmingly to the H2O cluster. The large peak in the photolysis cross-section spectrum of NaOH, σ(231.2 nm)=(18.2±0.18)×10−18 cm2, was reduced to (−0.13±0.15)×10−18 cm2, i.e. effectively zero. In contrast, σ(248.4 nm) changed from (1.73±0.13)×10−18 cm2 to (0.361±0.044)×10−18
cm−2. These changes would be accounted for if the NaOH spectrum was shifted to shorter wavelengths by the equivalent of 75 kJ mol−1, which is consistent with the NaOH–H2O binding energy. If the NaHCO3 photolysis spectrum is also shifted to shorter wavelengths by the NaHCO3–H2O binding energy, as predicted by theory, then the lifetime against photolysis in the mesosphere would increase from 2.2 to 4.5 h.
In the case of NaHCO3, the CIS excited state calculations show that the vertical transitions with large oscillator strengths (f>0.02) that contribute to the absorption band between 210 and 230 nm all lead to dissociation to Na and the HCO3 radical (Fig. 3). It has been speculated that HCO3 could be formed in the atmosphere from the reaction
| | 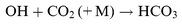 | (R7) |
However, the present calculations reveal two significant features on the potential energy surface of reaction (
R7). First, the complex is weakly bound, Δ
H0°(7)
=
−14
±
20 kJ mol
−1. Second, there is a significant barrier Δ
E0#=
74 kJ mol
−1
for HCO
3 to dissociate to OH+CO
2. Thus, reaction (
R7) is likely to be extremely slow. On the other hand, HCO
3 will also not dissociate very rapidly. We have estimated the rate of thermal dissociation using Rice–Ramsberger–Kassel–Marcus (RRKM) theory
31 with the data in
Table 10. This indicates that at a pressure of 0.01 Torr (the pressure at 80 km in the atmosphere), the rate of dissociation of HCO
3 is 1.4
×
10
8 exp(−8400/
T) s
−1. Even allowing for the expected uncertainty
16 of ±14 kJ mol
−1 in the calculated barrier height, HCO
3 should be thermally stable for at least several days at 80 km, and is likely to be destroyed much more rapidly by reaction
with atomic O or H.
Table 10 Calculated molecular parameters at the B3LYP/6-311+G(2d,p) level of theory for the minimum and saddle point on the OH+CO2→HCO3 surface
| Species (see Fig. 3) |
Dipole momenta |
Rotational constantsb |
Vibrational frequenciesc |
ΔH0od |
| |
|
In D≈3.33564×10−30 C m.
In GHz.
In cm−1.
In kJ mol−1, with respect to OH +CO2
(zero-point energies included).
|
| HCO3 |
2.84 |
13.8, 11.3, 6.20 |
458, 514, 556, 760, 1031, 1149, 1278, 1565, 3757 |
−13.5 |
| HO–CO2 saddle point |
2.17 |
13.7, 9.19, 5.59 |
569(i), 330, 395, 572, 675, 901, 1235, 2104, 3728 |
60.7 |
Acknowledgements
This work, and a research studentship for DES, were supported by the Natural Environment Research Council (grant GR3/11754).
References
- J. M. C. Plane, Int. Rev. Phys. Chem., 1991, 10, 55 CrossRef CAS.
- S. G. Love and D. E. Brownlee, Science, 1993, 262, 550 CrossRef.
- J. M. C. Plane, C. S. Gardner, J. Yu, C. Y. She, R. R. Garcia and H. C. Pumphrey, J. Geophys. Res., 1999, 104, 3773 CrossRef CAS.
- R. M. Cox, D. E. Self and J. M. C. Plane, J. Geophys. Res., 2001, 106, 1733 CAS.
- W. J. McNeil, E. Murad and S. T. Lai, J. Geophys. Res., 1995, 100, 16847 CrossRef CAS.
- R. J. States and C. S. Gardner, J. Geophys. Res., 1999, 104, 11783 CrossRef CAS.
- J. M. C. Plane, Ann. Geophys., 2000, 18, 807 Search PubMed.
- J. A. Silver, D. R. Worsnop, A. Freedman and C. E. Kolb, J. Chem. Phys., 1986, 84, 4378 CrossRef CAS.
- B. Rajasekhar, J. M. C. Plane and L. Bartolotti, J. Phys. Chem., 1989, 93, 7399 CrossRef CAS.
- J. M. C. Plane and B. Rajasekhar, J. Phys. Chem., 1989, 93, 3135 CrossRef CAS.
- A. Gaupp, P. Kuske and H. J. Andra, Phys. Rev. A, 1982, 26, 3351 CrossRef CAS.
- J. M. C. Plane, C.-F. Nien and B. Rajasekhar, J. Phys. Chem., 1992, 96, 1296 CrossRef CAS.
- R. W. Huggins and J. H. Kahn, J. Appl. Phys., 1967, 38, 180 CrossRef CAS.
- M. Helmer and J. M. C. Plane, J. Geophys. Res., 1993, 98, 23207 CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.7, Gaussian, Inc., Pittsburgh, PA, 1998. Search PubMed.
-
J. B. Foresman and A. Frisch, Exploring Chemistry with Electronic Structure Methods, Gaussian, Inc., Pittsburgh, PA, 1996. Search PubMed.
- R. M. Cox and J. M. C. Plane, Phys. Chem. Chem. Phys., 1999, 1, 4713 RSC.
- J. M. C. Plane, C.-F. Nien, M. R. Allen and M. Helmer, J. Phys. Chem., 1993, 97, 4459 CrossRef CAS.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery, Jr., J. Chem. Phys., 1996, 104, 2598 CrossRef CAS.
- S. R. Langhoff, H. Partridge and C. W. Bauschlicher, Jr., Chem. Phys., 1991, 153, 1 CrossRef CAS.
- J. V. Pugh, K. K. Shen, C. B. Winstead and J. L. Gole, Chem. Phys., 1996, 202, 129 CrossRef CAS.
- A. Ellis, Personal communication..
- T. G. Wright, A. M. Ellis and J. M. Dyke, J. Chem. Phys., 98, 2891 Search PubMed.
- S. Joo, D. R. Worsnop, C. E. Kolb, S. K. Kim and D. R. Herschbach, J. Phys. Chem., 1999, 103, 3193 Search PubMed.
- L. Andrews, J. Mol. Spectrosc., 1976, 61, 337 CrossRef CAS.
- F.
S. Rowland and Y. Makide, Geophys. Res. Lett., 1982, 9, 473 CAS.
- U. A. Gottwald, P B. Monkhouse and B. Bonn, Appl. Phys. B, 1999, 69, 151 CrossRef CAS.
- J. C. Arvensen, R. N. Griffin and B. D. Pearson, Jr., Appl. Opt., 1969, 8, 2215.
- J. Griffin, D. R. Worsnop, R. C. Brown, C. E. Kolb and D. R. Herschbach, J. Phys. Chem., 2001, 105, 1643 Search PubMed.
- D. M. Hunten, R. P. Turco and O. B. Toon, J. Atmos. Sci., 1980, 37, 1342 Search PubMed.
-
R. G. Gilbert and S. C. Smith, Theory of Unimolecular and Recombination
Reactions, Blackwell, Oxford, 1990. Search PubMed.
- R. G. Roble and E. C. Ridley, Geophys. Res. Lett., 1994, 21, 417 CrossRef CAS.
|
| This journal is © the Owner Societies 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.