Morphine, the Proteus of organic molecules
Paul R.
Blakemore
a and
James D.
White
*b
aSchool of Chemistry, University of Leeds, Leeds, UK LS2 9JT
bDepartment of Chemistry, Oregon State University, Corvallis, Oregon 97331-4003, USA
First published on 14th February 2002
Abstract
This feature article encapsulates the senior author’s long-standing interests in opiate chemistry and attempts to place it within an historical context and against the backdrop of related work by others who have viewed morphine as one of the pinnacles of natural product synthesis. Biomimetic and ‘bioanalogous’ routes to the morphine skeleton are discussed followed by approaches based on the elaboration of phenanthrene platforms. The latter include an asymmetric synthesis of ent-morphine developed in our laboratory.
Paul R. Blakemore was born in 1973 in Dagenham, Essex, and received his BSc degree from Southampton University in 1995. He completed graduate work with Professor P. J. Kocienski at the University of Glasgow, and after receiving his PhD degree in 1999, held a postdoctoral appointment at Oregon State University with Professor J. D. White until autumn 2001. He recently returned to the UK to take up a Royal Society University Research Fellowship at the University of Leeds. |
James D. White was born in Bristol and received his early education in India and in Tiverton, Devon. After National Service as a Pilot Officer in the RAF, he went up to Queens’ College, Cambridge, and received his BA degree in 1959. He began postgraduate studies at the University of British Columbia under Raymond Bonnett, obtaining the MSc degree in 1961, then moved to the Massachusetts Institute of Technology where he completed his PhD in 1965 with the late George Büchi. His first academic appointment was at Harvard University as Instructor and then Assistant Professor, and in 1971 he moved to his present location in Corvallis as Associate Professor of Chemistry at Oregon State University. Currently, he holds the rank of Distinguished Professor at that University. His research has embraced many of the sub-disciplines of natural products chemistry, including terpenoids, alkaloids, macrolides, depsipeptides, and eicosanoids, and he continues to pursue synthetic studies in these areas alongside active participation on the senior tennis circuit. |
Introduction
‘Of all the remedies which a kind Providence has bestowed upon mankind for the purpose of lighting its miseries, there is not one which equals opium in its power to moderate the violence of so many maladies and even to cure some of them.’Thomas Sydenham, 1624–1689.
The powerfully euphoric and analgetic opium alkaloids have fascinated mankind since the days of antiquity. The influence of the opiates on modern society cannot be over-estimated; they are used extensively as medicines to ease human suffering and are abused in equal measure as illicit narcotics. Annual global production of opium by cultivation of the readily grown poppy Papaver somniferum is enormous. In the year 2000, an estimated 8700 tons† of opium and poppy straw concentrate were produced worldwide from over 300000 hectares‡ of poppy fields.1 The active principle of opium, and the motivation behind such industry, is morphine (1), a molecule which can lay claim to being the original alkaloid and the first true drug. Early attempts to unlock the mysteries of opium provided a major stimulus to the development of organic chemistry and, it may be argued, spawned the entire field of medicinal chemistry. Indeed, Sir Robert Robinson referred to morphine in affectionate terms as, ‘this veritable Proteus among molecules.’2
Synthetically inclined chemists have long pondered the complex challenge associated with assembling the morphine scaffold. Gates completed his landmark synthesis of morphine in 1952,3 and since that time many have attempted its betterment, some with more success than others. Numerous reviews4 and treatises5 of morphinan chemistry have appeared which make good attempts at covering this vast subject. The present article is not intended to rival those works. Rather, it endeavours to trace the development of our own efforts towards the synthesis of morphine over the past twenty years and to set it against the broad tapestry of work in the opiate field.
History, production and biological action of morphine
The opium poppy, Papaver somniferum, probably originated in Asia Minor and there are strong indications that the Sumerians cultivated it to extract opium at least three thousand years before Christ.6 Late bronze age Cypriote artifacts from ca. 1500 BC have been found which are evocatively stylized to resemble poppy seed capsules. In The Odyssey the Greek poet Homer spoke of the poppy being ‘saturated with lethal slumber.’ Poppy cultivation for opium production spread inexorably around the world, and from the fourth century BC onwards opium became widely recognised as an analgesic of unrivalled power. Two thousand years later, in the sixteenth century, the Swiss alchemist Paracelsus introduced Europe to laudanum, an alcoholic tincture of opium and an assortment of other exotic ingredients which included ambergris and musk.7 The English physician Thomas Sydenham (1624–1689) later improved upon the recipe and noted that it was particularly good for plague. Laudanum was a cornerstone of European medicine until well into the nineteenth century and had its fair share of abusive users, most famously the essayist De Quincey.8The modern era of narcotics arrived in 1806 when Sertürner isolated the active principle of opium in pure form.9 He named the white crystalline powder morphinum, after Morpheus, the Greek god of dreams. Sertürner observed that morphinum, or morphine as we now know it, belonged to an as yet unknown class of natural products, ‘the vegetable alkalis’ or alkaloids. The purified alkaloid elicited a greater biological response than opium-based concoctions, and its use, both legitimate and otherwise, proliferated with the invention of the hypodermic syringe in 1853 which provided a superior method of administration. Later research by Heinrich Dreser at Friedrich Bayer and Company led to the discovery of diacetylmorphine (diamorphine). This early synthetic drug was marketed to the German people in 1898 as a cough remedy under the brand name Heroin (derived from the German word for heroic). Although diacetylmorphine is metabolised to morphine in the body, it has the ability to rapidly cross the blood-brain barrier and create an euphoric ‘rush’. This property made the drug particularly addictive and led to an immense illicit demand for heroin which continues to this day. Codeine (2), the biogenetic precursor of morphine and also a constituent of opium, has a more tempered biological action than either morphine or heroin and is commonly prescribed as an antitussive agent and mild analgesic.
Robinson proposed the correct structure for morphine in 1925,10 although structural investigations of the alkaloid had begun soon after its initial isolation by Sertürner. Early studies by Liebig were followed by those of Laurent, who correctly deduced the empirical formula for morphine as C17H19NO3 in 1847.11 Robinson’s keen insights were built not only on the results of these and later pioneers in the morphine area (chief among them Knorr and Pschorr), but also on his own extensive degradation experiments.12 Gates confirmed the constitutional accuracy of Robinson’s morphine structure by total synthesis in 1952,3 and in doing so realised a long standing goal of synthetic organic chemistry. Ever since the seminal work of Sertürner, great interest in a totally synthetic preparation of morphine had existed. After all, if morphine could be synthesised, then why not analogous compounds with perhaps less nefarious side-effects? To promote this goal, the Prussian Academy of Science in 1870 reputedly offered one hundred ducats to any individual who could synthesise morphine, and later in 1925 a wealthy American manufacturer offered $100000 for the same purpose. Gates and his co-worker Tschudi were unfortunately too late to pocket either of these prizes!
A genuinely practical synthetic route to morphine, i.e. one that can compete with the natural supply, remains elusive. This is not surprising when one considers the comparative ease with which the opium poppy can be cultivated against the complexity of the target molecule in question. An average Indian acreage§ of P. somniferum yields between 25 and 30 kg of opium per season and, after refining, this will afford approximately 3 kg of morphine (or 300000 standard medical units).13 Extraction of opium from Papaver somniferum is a fairly simple affair and little has changed in the harvesting method since antiquity. Around fifteen days after petal fall, the immature seed capsule of the poppy plant is lanced and the fresh pink opium latex is exuded (Fig. 1). The next day, the latex, which by now is black, is scraped from the capsule and collected. Each capsule is lanced a further three or four times over the following week and yields more of its precious crop. The raw opium is partially dehydrated by sun baking to remove about 90% of its water content; at this point, the black resinous mass is known commercially as Indian opium and contains approximately 10-15% morphine (1), 3–4% codeine (2), 1–7% narcotine, 0.5–1% papaverine, and 0.1–2% thebaine (3). Further processing of the opium yields purified morphine, the majority of which is used to manufacture codeine by simple phenolic methylation. New varieties of Papaver somniferum are now being grown in Australia which produce greater quantities of thebaine, a biogenetic precursor of morphine and codeine. Thebaine is vital for the production of synthetic opiates such as oxymorphone (4), naloxone (5), and etorphine (6).
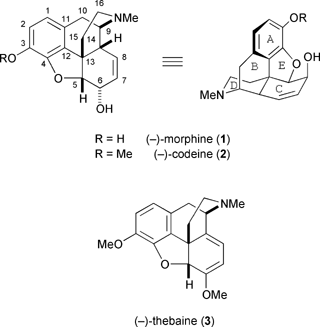
 | ||
| Fig. 1 A lanced poppy seed capsule exuding opium latex. | ||
The opiates elicit their intense biological response by binding to specific receptor sites within the central nervous system.14 Three distinct classes of mammalian opioid receptors, μ, κ, and δ, have been identified, and there is evidence that these may be further divided into various sub-types. These transmembrane receptor proteins are coupled to Gi proteins and thereby trigger the inhibition of adenylyl cyclase, the enzyme responsible for cyclic adenosylmonophosphate (cAMP) production. The attendant lowering of cAMP levels affects the potassium (δ and μ receptor agonism) and calcium (κ receptor agonism) ion channels of the cell. If use of an opiate drug is interrupted, there is a sudden surge in cellular cAMP production which precipitates a multitude of serious withdrawal symptoms.
It was long suspected that morphine was not a natural opioid receptor ligand, but that it mimicked some as yet undiscovered endogenous analgesic compound(s). Hughes was among the first to confirm this hypothesis by isolating the peptidic enkephalins from mammalian brain tissue in 1975.15 The two enkephalins, met-enkephalin (Tyr-Gly-Gly-Phe-Met) and leu-enkephalin (Tyr-Gly-Gly-Phe-Leu), were characterized by their ability to inhibit the electrically stimulated contraction of guinea pig ileum and mouse vas deferens. Morphine similarly inhibits such contractions. The enkephalins share 80% homology with each other, and an obvious similarity exists between tyrosine, the first amino acid residue of each, and morphine. Other peptidic endogenous opioid receptor ligands were soon found; these peptides are now collectively known as endorphins, a shortened form of endogenous morphinoids.16 Endorphins are produced by the body in response to states of extreme stress and pain. For instance, it may be demonstrated that the analgetic effect of acupuncture is due to stimulated endorphin release.
The structure–activity relationships (SAR) for morphine analogues have been extensively mapped.17 Minor modification of the morphine nucleus can yield opioids with altered receptor affinities and fundamentally different pharmacologies. Morphine itself is a selective μ-type receptor agonist, and so the analgesia it elicits is also accompanied by euphoria, decreased gastrointestinal motility (leading to constipation), physical dependence, and respiratory depression. The last is usually the cause of death upon overdose. Synthetic oxymorphone (4) is a μ-agonist with ten times the potency of morphine, but the closely related naloxone (5) actually antagonises all types of opioid receptors and is a useful antidote to morphine overdosage. Etorphine (6) has a thousand times the potency of morphine as a μ-agonist but also greatly depresses respiration. Its principal use is reserved for the immobilisation of very large animals, such as elephants and rhinos. Agonism of κ-type receptors also elicits analgesia but without the respiratory depression, euphoria, and constipation associated with μ-agonism. Selective κ-agonists have been identified as potential therapeutic agents, but unfortunately they have strong sedative and dysphoric side effects.
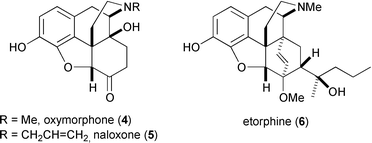
Many opioids (i.e. compounds which possess opioid receptor affinity) have been discovered, by both systematic research and serendipity, which bare only a very slight structural resemblance to morphine and other true opiates.17 For instance, meperidine (7) was originally conceived as an antispasmodic agent18 but was later found to be an effective μ-type agonist. Meperidine (Demerol) has about one tenth the analgesic strength of morphine but is less constipating and does not suppress the cough reflex. Methadone (8) was discovered by German scientists during the second world war, and again was intended for use as an antispasmodic. It is employed today as a long-acting, orally-administered analgesic for cancer sufferers and also to treat heroin addicts. Sufentanil (9) is one of a growing family of 4-anilidopiperidine (fentanyl) analgesics, and is 600 to 800 times as potent as morphine. Despite its greater analgetic strength, sufentanil does not significantly depress respiration and finds use as an anesthetic for surgical procedures.
Biosynthesis of morphine
A detailed understanding of the biogenesis of morphine from L-tyrosine in P. somniferum is now almost complete (Scheme 1).19 During the biosynthesis, L-tyrosine (10) is effectively ‘dimerised’ and provides the source for all of the non-methyl carbon atoms incorporated into morphine. One of the tyrosine equivalents is converted into dopamine (11) in two enzymatic steps, either viaL-DOPA or tyramine, depending on whether aromatic hydroxylation is followed by decarboxylation or vice versa. The action of a transaminase on the second equivalent of tyrosine yields p-hydroxyphenylpyruvic acid which is subsequently decarboxylated to give p-hydroxyphenylacetaldehyde (12). The two halves of the embryonic morphine nucleus, 11 and 12, are now connected by a stereospecific Pictet-Spengler-type reaction mediated by (S)-norcoclaurine synthase. The product from this process, (S)-norcoclaurine (13), subsequently undergoes oxidation and methylation to yield (S)-reticuline (14). Interestingly, this substrate is not transformed into (+)-morphine (a compound unknown in the natural world), but is first converted into its enantiomer, (R)-reticuline (15), the precursor to (−)-morphine. The inversion of configuration occurs by an oxidation–reduction sequence in which an iminium species generated from 14 by (S)-reticuline oxidase is stereospecifically reduced to (R)-reticuline with the NADPH-requiring enzyme 1,2-dehydroreticulinium reductase.20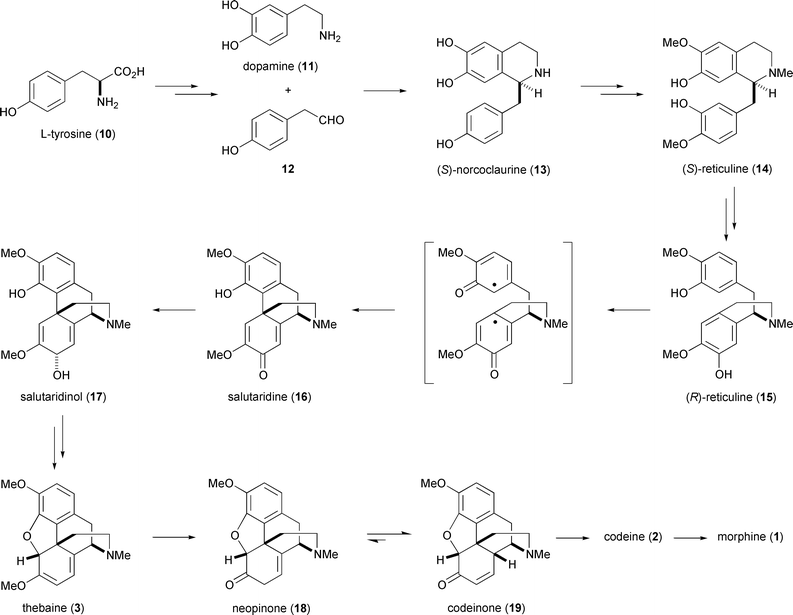 | ||
| Scheme 1 Biosynthesis of morphine. | ||
The next biogenetic step, conversion of (R)-reticuline to salutaridine (16) occurs by way of a regioselective oxidative phenolic coupling. A NADPH-dependent cytochrome P450, salutaridine synthase, is responsible for this remarkable transformation. Unlike the cytochrome P450s commonly implicated in alkaloid biosynthesis, salutaridine synthase is an oxidase rather than a mono-oxygenase, i.e. no atoms from molecular oxygen are incorporated into the product. Further aspects of this prototypic phenolic coupling and its relevance to synthetic strategies for morphine synthesis will be discussed in the next section.
The keto group of salutaridine (16) is reduced by salutaridine reductase to yield the (7S) alcohol salutaridinol (17),21 and the latter is then acetylated by salutaridinol 7-O-acetyltransferase. The resulting allylic acetate suffers a spontaneous non-enzymatic syn SN2’ displacement by the phenolic hydroxy group to create the fifth and final ring of the morphine scaffold. The product from this cyclisation event is thebaine (3), which is demethylated to yield neopinone (18) by an as yet uncharacterised enzyme. Neopinone readily isomerizes to codeinone (19) which is duly reduced to codeine (2) by codeinone reductase. Final demethylation of codeine yields morphine (1) which is itself subject to further metabolic transformation. The enzyme which catalyses the demethylation of codeine has not yet been characterised in P. somniferum, but the reaction has been mapped in mammalian liver.22
Biomimetic and ‘bioanalogous’ approaches to morphine synthesis
The most interesting step in the biogenesis of morphine is undoubtedly the diphenolic coupling which forges the linkage between the two aromatic rings of (R)-reticuline and leads to salutaridine. Barton and Cohen originated the idea that coupling reactions of this type underlay the formation of many C–C and C–O bonds in a variety of alkaloids, including morphine.23 Barton’s curiosity about these intriguing reactions was aroused by his rejection of the then commonly accepted structure for ‘Pummerer’s ketone’, a dimeric oxidation product of p-cresol (20).24 Barton correctly deduced the structure of the dimer as 21 rather than 22, and he provided a plausible mechanistic rationale for its formation (Scheme 2).25 An obvious similarity exists between dienone 23 and salutaridine (16), and Barton was quick to recognise the significance of his conclusions about Pummerer’s ketone to morphine biosynthesis.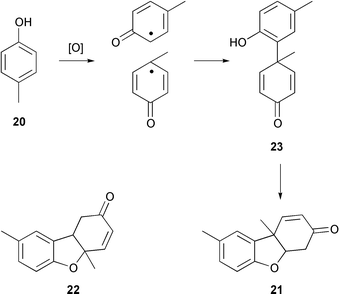 | ||
| Scheme 2 Formation of Pummerer’s ketone (21). | ||
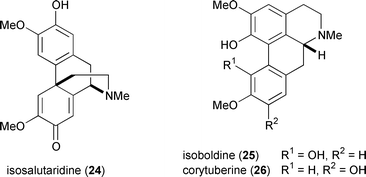
Anxious to lend credence to his hypothesis for morphine biosynthesis, Barton demonstrated the in vitro conversion of isotopically labeled reticuline (15) to salutaridine (16) by treatment with potassium ferricyanide.26 A dilution experiment determined the yield for this transformation at a barely discernible 0.02%! Later, Schwartz improved upon this biomimetic conversion by replacing the ferricyanide oxidant with thallium tris(trifluoroacetate).27 Despite numerous efforts at optimising the coupling process, however, the yield of salutaridine has remained disappointingly low. The non-enzymatically mediated oxidative coupling of reticuline suffers from inherently low regioselectivity and several products are formed, including significant quantities of isosalutaridine (24), isoboldine (25), and corytuberine (26).
Despite the difficulties associated with achieving an efficient non-enzymatic conversion of reticuline to salutaridine, the concept provided fertile ground for a number of approaches to morphine by Schwartz and others.28 We became preoccupied with this challenging problem in the early 1980’s29 and reasoned, as had Szántay,30 that a hypervalent iodine-based oxidant31 would improve the coupling process. Our hope was that without the intervention of radical intermediates that attend one-electron phenolic oxidations such as those carried out with ferricyanide, for example, a less complex mixture of products would ensue.32 Heterolytic bond formation and scission, it was believed, would offer a mild solution to the oxidative coupling problem which could perhaps circumvent the formation of unwanted (for morphine synthesis, at least) substances such as 24, 25, and 26.
After screening a variety of iodine(III) oxidants, phenyliodoso bis(trifluoroacetate)33 was identified as the preferred reagent for the conversion of N-trifluoroacetyl-6’-bromonorreticuline (27) to the salutaridine derivative 28 (Scheme 3). Unfortunately, the new oxidation method proved little better than traditional procedures, and despite the use of brominated reticuline derivative 27 to prevent para–para coupling,34 the salutaridine derivative 28 was isolated in only 21% yield. Nevertheless, 28 was successfully converted into (–)-codeine (2) in a relatively short sequence of reactions. Saponification of the trifluoroacetamide was followed by methylation of the liberated amine, and concomitant reduction of the ketone yielded a 1∶1 mixture of α- and β-bromosalutaridinols. Activation of either of the stereoisomeric alcohols with the dineopentyl acetal of dimethylformamide gave 1-bromothebaine (29) in a SN2’ process which closely resembles the analogous biochemical transformation. Hydrolysis of the enol ether to 29 followed by reduction of both the enone and aryl bromide of 30 completed the synthesis of (−)-codeine (2) and thus a formal synthesis of (−)-morphine (1).
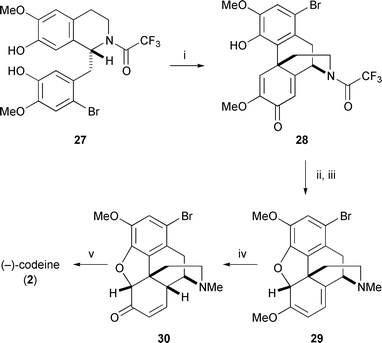 | ||
| Scheme 3 Reagents and conditons: i, PhI(OCOCF3)2, CH2Cl2, −40 °C, 21%; ii, aq. K2CO3, MeOH, then aq. CH2O, then NaBH4, 68%; iii, Me2NCH(OCH2CMe3)2, CH2Cl2, 80%; iv, Hg(OAc)2, HCO2H, then HCl, Et2O; v, LiAlH4, THF, Δ, 25% from 29. | ||
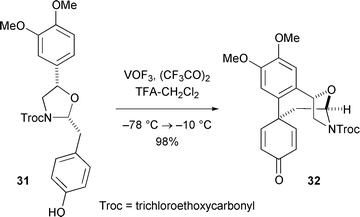
The disappointing outcome of most oxidative phenolic couplings carried out on reticuline derivatives persuaded us that, with suitable constraints in place, intramolecular reactions of this type could be made to occur with greater efficiency. This led to the notion that an oxazolidine could serve as an effective template upon which directed phenolic oxidative coupling would take place if aryl substituents were correctly oriented.35 Indeed, treatment of chiral oxazolidine 31 with vanadium oxytrifluoride36 gave the para–para coupling product 32 stereospecifically and in near quantitative yield. Phenyliodoso bis(trifluoroacetate) also effected this transformation, but with reduced yield. Although the coupling mode illustrated here leads exclusively into the isosalutaridine manifold, further transformations carried out with 32 suggest that the principle intrinsic to the template controlled phenolic coupling of 31 could be exploited profitably in an approach to morphine.
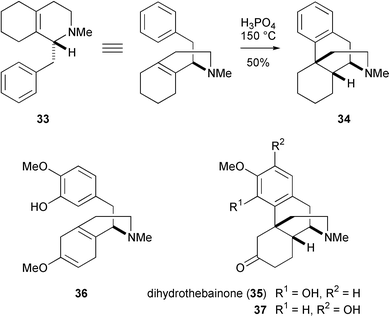
As outlined above, biomimetic oxidative phenolic coupling of reticuline derivatives have proved to be of limited value for the preparation of morphine and its congeners in the absence constraining factors such as those present in 31. However, formation of the same C12–C13 bond by alternative means has led to efficient strategies for opiate synthesis. Grewe developed one of the first ‘bioanalogous’ approaches to morphinans in 194837 by heating the 1-benzyloctahydroisoquinoline derivative 33 in phosphoric acid. These conditions initiated a cationic cyclisation to the morphinan skeleton 34 in remarkably good yield. Some twenty years later, Grewe extended this protocol to achieve a formal synthesis of morphine via dihydrothebainone (35).38 In that instance, however, a regioselectivity problem seriously detracted from the efficiency of cyclisation, since the enol ether 36 upon treatment with phosphoric acid gave predominantly the unwanted product 37 of para coupling (37%) rather than the ortho coupling product 35 (3%). Morrison independently arrived at the same disappointing outcome,39 but Beyerman cleverly avoided the regioselectivity problem of the Grewe cyclisation by employing a symmetrically substituted benzyl moiety. A temporary hydroxy substituent at C2 (morphine numbering) provided the local symmetry and was later removed for the final convergence on dihydrothebainone.40
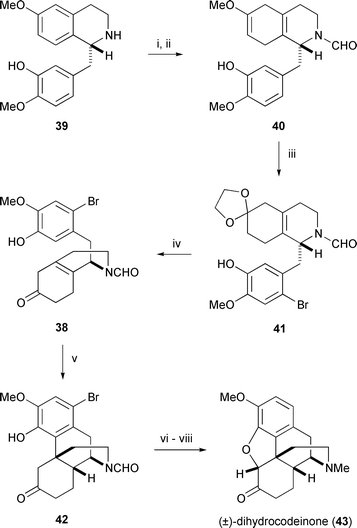
The Grewe cyclisation has formed the basis of the most practical synthesis of morphine to date. In a remarkably straightforward route, Rice employed a bromine substituent as a blocking group at C1 (morphine numbering) of the Grewe cyclisation precursor 38 to prevent para coupling (Scheme 4).41 Birch reduction of the tetrahydroisoquinoline 39, derived from 3-methoxyphenethylamine in two steps, left the more electron-rich catechol ring unscathed and gave amide 40 after formylation. Ketalisation and regioselective bromination of 40 was carried out consecutively in a single reaction vessel, and after a simple work-up the crude ketal 41 was immediately hydrolysed. Grewe cyclisation of 38 was initiated by hydrogen fluoride–ammonia complex in triflic acid, and after 3–4 days at 0 °C afforded a 60% yield of 1-bromo-N-formylnordihydrothebainone (42). Bromination α to the ketone derived from amide hydrolysis of 42 was followed by base-induced ring closure to complete the pentacyclic framework. Cleavage of the aryl bromide and methylation of the secondary amine were achieved concomitantly by hydrogenation over palladium in a mixture of acetic acid and formalin and led to racemic dihydrocodeinone (43).
 | ||
| Scheme 4 Reagents and conditions: i, Li, THF–t-BuOH–NH3, −55 °C, 90%, ii, PhOCHO, EtOAc, Δ, 94%; iii, cat. MsOH, ethylene glycol, THF, then NBS; iv, HCO2H–H2O, 90% from 40; v, NH4F·HF, TfOH, 0 °C, 60%; vi, aq. HCl, MeOH, Δ; vii, Br2, AcOH, then aq. NaOH, CHCl3; viii, H2, 10% Pd/C, NaOAc, aq. CH2O, AcOH, 79% from 42. | ||
Rice’s synthesis of dihydrocodeinone was further modified to yield dihydrothebainone (35) and nordihydrocodeinone, compounds that are important precursors to natural opiates including morphine and thebaine-based drugs such as naloxone (5) and etorphine (6). It is noteworthy that the entire Rice synthesis of dihydrocodeinone (43) involves the isolation of only six intermediates, requires no chromatographic purifications, and proceeds in 29% overall yield!
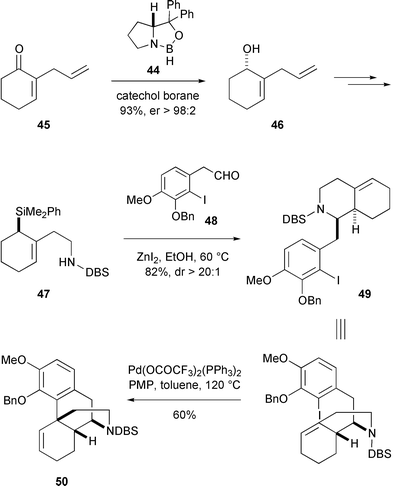
It is appropriate to conclude this survey of biomimetic and ‘bioanalogous’ approaches to morphinans, with a brief description of the first genuinely asymmetric synthesis of morphine. Remarkably, all of the published syntheses of morphine until 1993 were either racemic or involved classical resolution. Overman rectified this situation by reporting a true asymmetric route which was almost certainly inspired by the earlier contributions of Grewe (Scheme 5).42The source of chirality for the Overman synthesis of morphine was provided by the oxazaborolidine reagent 44,43 which catalysed an asymmetric reduction of enone 45 by catecholborane. Standard synthetic operations on the allylic alcohol 46 then gave allyl silane 47. Condensation of the secondary amine function of the latter with aldehyde 48 produced an iminium ion intermediate which was trapped by the pendant allyl silane moiety to yield octahydroisoquinoline 49 with high diastereoselectivity. In the pivotal step of the synthesis, an intramolecular Heck reaction of 49 created the B-ring of morphine in a reaction which bears a notable resemblance to the classical Grewe cyclisation. The success of the Heck coupling confirmed Overman’s view that the intramolecular version of this reaction offers a method par excellence for setting congested quaternary stereogenic centres. The synthesis of (−)-morphine was formally completed by a further four steps which transformed the Heck cyclisation product 50 into (−)-dihydrocodeinone (43).44 The availability of the proline-derived catalyst 44 in both enantiomeric forms means, of course, that both (−)- and (+)-morphine can be prepared by this route.
 | ||
| Scheme 5 DBS = dibenzosuberyl, PMP = 1,2,2,6,6-pentamethylpiperidine. | ||
Phenanthrene-based approaches to morphine
Before the structure of morphine was fully elucidated by Robinson, synthetic studies were influenced almost entirely by its degradation chemistry. Many harsh degradative treatments of morphine resulted in aromatic phenanthrene derivatives, and the parent hydrocarbon became regarded as a ‘primeval’ form of the alkaloid. For example, it was well known that distillation of morphine with zinc dust yielded phenanthrene,45 and that exhaustive Hofmann degradation of codeine gave methylmorphenol 51.46 These experiments encouraged some of the first synthetic forays towards morphine even though there was no clear structural delineation of the target at the time.47 Indeed, Robinson himself later adopted an approach which hinged upon elaboration of the phenanthrene nucleus.48 To date, however, only three completed syntheses have appeared which fabricate the morphine molecule from a phenanthrene core. A phenanthrene based approach to morphine necessitates a challenging C13–C15 bond forming event to create the quaternary stereogenic centre at C13, whereas the more common bioanalogous routes discussed above achieve the same goal employing a C12–C13 bond construction.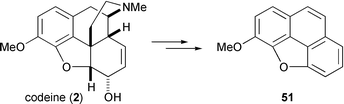
Ginsburg reported the first phenanthrene based route to morphine in 1954,49 just two years after Gates’ achievement3 (Scheme 6). The Ginsburg synthesis, like that of Gates, was accomplished without the benefit of many of the methods that are now a familiar part of the synthetic chemists’ armamentarium. Indeed, Ginsburg’s synthesis of morphine is a fitting reminder of what a skilled chemist can achieve with a set of rudimentary reactions, good planning, and an element of luck.
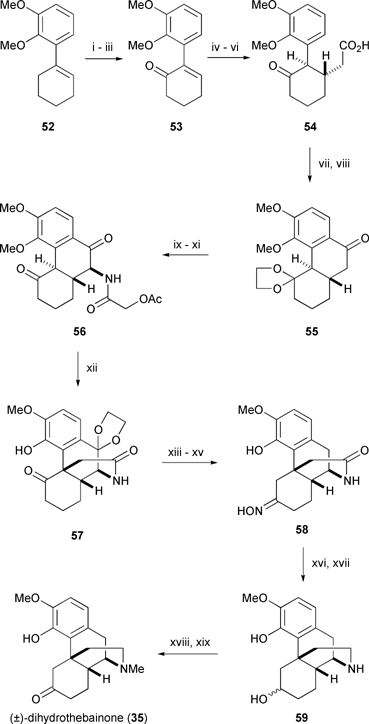 | ||
| Scheme 6 Reagents and conditons: i, n-C5H11ONO, AcOH, aq. HCl, −10 °C, 65%; ii, pyridine, 70 °C, 75%; iii, aq. H2SO4, Δ, 90%; iv, benzyl malonate, t-BuOK, t-BuOH, 60 °C; v, H2, Pd/C, AcOH; vi, 170 °C (−CO2), 88% (3 steps); vii, HF, 90%; viii, cat. TsOH, ethylene glycol, benzene, Δ; ix, NaOEt, n-C5H11ONO, EtOH–dioxane, then aq. AcOH, 76%; x, H2, Pd/C, aq. HCl, EtOH, 60%; xi, AcOCH2COCl, pyridine, CHCl3, Δ, 76%; xii, cat. TsOH, ethylene glycol, benzene–toluene, Δ; xiii, NaOEt, n-C5H11ONO, EtOH–dioxane, then aq. AcOH; xiv, aq. HCl, EtOH, Δ, 80%; xv, NH2NH2·H2O, diethylene glycol, 165 °C, 45%; xvi, aq. HCl, EtOH, Δ; xvii, LiAlH4, THF, Δ xviii, CH2O, HCO2H, Δ; xix, Ph2CO, t-BuOK, benzene, Δ. | ||
Ginsburg’s route to morphine began with addition of ortho-lithiated veratrole to cyclohexanone, followed by acid catalysed dehydration, to afford cyclohexene 52.50 An unusual allylic oxidation method — nitrosochlorination, followed by elimination, and oxime hydrolysis51 — furnished enone 53 which underwent a facile Michael reaction with dibenzyl malonate. After hydrolysis and decarboxylation of the resulting malonate ester, carboxylic acid 54 was cyclised to yield a diketo phenanthrene derivative which was selectively ketalised at C5 (morphine numbering) to yield dioxolane 55.52 α-Nitrosation of the free ketone was followed by hydrogenation, and subsequent acylation of the liberated amine gave the acetamide derivative 56.
An attempt to selectively ketalise the C5 carbonyl group of dione 56 initiated a remarkable series of reactions. Ketalisation occurred at C10 rather than C5 and the C4 phenolic ether was demethylated, but the most surprising outcome was loss of the elements of acetic acid and formation of the pivotal C13–C15 bond to yield morphinan 57.53 Rarely does an unanticipated transformation prove so profitable, and Ginsburg did not hesitate to take advantage of his good fortune.
Amyl nitrite was employed for a third and final time in this synthesis to effect oxime formation via nitrosation of ketone 57. Selective hydrolysis of the dioxolane of the resulting product yielded a diketooxime. The ketonic carbonyls of the latter were removed by a Wolff-Kishner reduction which furnished lactam oxime 58 in fair yield. The oxime moiety of 58 was hydrolysed, and following global reduction, the resulting alcohol 59 was N-methylated and oxidised to (±)-dihydrothebainone (35). At this point the route converged with Gates’ synthesis of morphine and was formally complete.
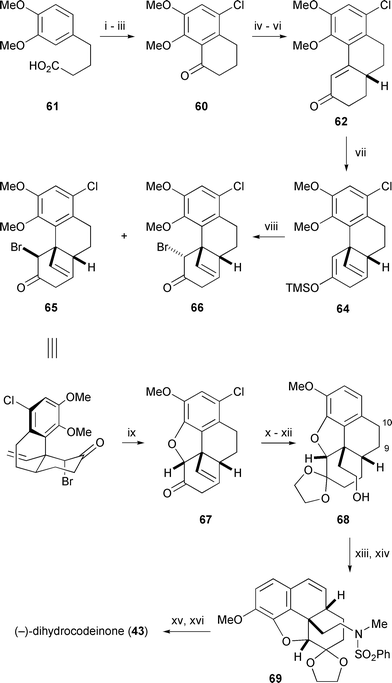
That the next successful phenanthrene based route to morphine did not appear for over forty years is indicative of the inherent difficulty of the approach and its consequent lack of popularity. It was left to Mulzer in Vienna and ourselves to take up the challenge almost simultaneously in the early 1990s. Mulzer followed a strategy which conceptually reversed the Hofmann degradation of morphine and relied upon conjugate addition of a cuprate to forge the critical C13–C15 bond (Scheme 7).54 In this approach, tetralone 60 was prepared in three steps from the commercially available carboxylic acid 61, where a chlorine substituent blocked an otherwise favorable intramolecular Friedel-Crafts acylation towards the undesired carbocycle. A Robinson annulation sequence converted 60 to the racemic phenanthrene derivative 62 which was resolved by chiral phase chromatography on cellose triacetate.
 | ||
Scheme 7
Reagents and conditions: i, Cl2, AcOH, 99%; ii, (COCl)2, benzene, Δ, iii, SnCl4, benzene, 0 °C, 71% (2 steps); iv, HCO2Me, NaOMe, benzene, 95%; v, methyl vinyl ketone, Et3N, MeOH; vi, KOH, dioxane–H2O, 81% (2 steps); vii, (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2CuMgCl, THF, −78 °C → 0 °C, then TMSCl, Et3N, 0 °C → 25 °C; viii, NBS, THF, 81%, 65∶66 = 74∶26; ix, DMF, 140 °C, 99%; x, TMSCl, ethylene glycol, CH2Cl2, 92%; xi, BH3·SMe2, THF, then aq. H2O2, NaOH, 70%; xii, Raney Ni, KOH, MeOH, 98%; xiii, PhSO2NHMe, ADDP, Bu3P, benzene, 81%; xiv, (BzO)2, NBS, CCl4, Δ then Et3N, 67%; xvi, Li, NH3, THF–t-BuOH, −78 °C,
79%; xv, aq. HCl, 90 °C, 95%. CH)2CuMgCl, THF, −78 °C → 0 °C, then TMSCl, Et3N, 0 °C → 25 °C; viii, NBS, THF, 81%, 65∶66 = 74∶26; ix, DMF, 140 °C, 99%; x, TMSCl, ethylene glycol, CH2Cl2, 92%; xi, BH3·SMe2, THF, then aq. H2O2, NaOH, 70%; xii, Raney Ni, KOH, MeOH, 98%; xiii, PhSO2NHMe, ADDP, Bu3P, benzene, 81%; xiv, (BzO)2, NBS, CCl4, Δ then Et3N, 67%; xvi, Li, NH3, THF–t-BuOH, −78 °C,
79%; xv, aq. HCl, 90 °C, 95%. | ||
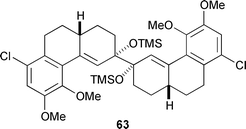
A survey of various conditions for the cuprate addition to enone 62 which established the C13 quaternary centre proved to be particularly fruitful. The usual cuprate addition protocol in which trimethylsilyl chloride is present to accelerate reaction led to significant quantities of a C2 symmetric pinacol dimer 63. However, when the Gilman-type cuprate derived from vinylmagnesium chloride and copper(I) iodide was added to enone 62followed by trimethylsilyl chloride, the pinacol coupling product was absent. The silyl enol ether 64 resulting from the cuprate addition was brominated to give a ca 3∶1 mixture of diastereomeric bromides 65 and 66, respectively. The X-ray crystal structure of 65, as well as its solution phase conformation determined by NMR analysis, revealed that this bromo ketone exists in a conformation (illustrated) well suited for subsequent cyclisation,55 and in fact, mere heating of 65 initiated an unusual internal etherification which gave morphenol derivative 67 directly in quantitative yield. The minor isomer 66 was found to have a very different conformation from 65 and was unsuitable for cyclisation. However, it could be recycled to 64 by reduction with zinc dust in the presence of trimethylsilyl chloride.
With the key step behind him, Mulzer completed his synthesis without incident. A straightforward sequence converted the vinyl moiety of 67 into a substituted ethylamine and introduced unsaturation between C9–C10. Removal of the sulfonamide from 69 under dissolving metal conditions resulted in concomitant cyclisation at C9 to set in place the final piperidine ring. This interesting transformation has direct precedent in a serendipitous discovery by Parker during the course of her own morphine synthesis.56 Final hydrolysis of the dioxolane yielded (−)-dihydrocodeinone (43) and formally completed the Mulzer synthesis of morphine. An important lesson from Mulzer’s route to morphine is that careful conformational analysis of a valuable intermediate about to be surrendered to an uncertain fate can lend confidence and plausibility to an idea which might otherwise be eschewed. This principle is further illustrated in our own phenanthrene-based approach to morphine.
It was in 1993 when our interest in a morphine synthesis was rekindled by a chance encounter with some carbene chemistry originating in our studies of metal-catalyzed decomposition of diazoketones.57 A revisitation of the (at that time) long neglected phenanthrene route to morphine seemed an attractive prospect, given the enormous advances in synthetic methodology since Ginsburg’s publication in 1954.49 An intramolecular carbenoid C–H insertion58 was selected as the means for constructing the pivotal C13–C15 bond, and the phenanthrene platform upon which this reaction was to be fashioned became our first objective. That the strategy ultimately succeeded and provided an asymmetric route to the unnatural dextrorotatory enantiomer of morphine (ent-1) confirmed the view that, in spite of a half century of effort, new and novel approaches to morphine have yet to be discovered.59
Our journey began with a catalytic asymmetric hydrogenation of the cinnamate 70, derived from Stobbe condensation of isovanillin with dimethyl succinate. This reaction, which introduced the first stereogenic centre of the morphine nucleus (Scheme 8), directed all subsequent stereochemical events, the absolute configuration of the rhodium ligand MOD-DIOP (71)60 used in the hydrogenation dictating the enantiomeric series entered by this route. The early introduction of a branch point in a synthesis which allows equally facile access to both enantiomers of the target through a single reaction, rather than through resolution or chromatographic separation, presents a distinct advantage, and is a characteristic of both Overman’s morphine synthesis and our own.
![Reagents and conditions: i, [RhCl(COD)]2, MOD-DIOP ligand 71, H2, MeOH, 100%, er = 97∶3, ii, Br2, AcOH, 93%; iii, MsOH, P2O5, 75%; iv, H2, Pd/C, NaHCO3, MeOH, 100%; v, LiOH, THF–H2O, 100%; vi, KH, HCO2Me, DME, 0 °C, 85%; vii, methyl vinyl ketone, Et3N, CH2Cl2, 95%; viii, NaOH, H2O–THF, 95%; ix, CH2N2, Et2O–CH2Cl2, 99%; x, Br2, NaHCO3, CH2Cl2, 80%; xi, DBU, benzene, 50 °C, 90%; xii, NaBH4, i-PrOH–CH2Cl2, 99%; xiii, H2, Pd/C, MeOH, 78%, dr = 22∶1, xiv, CH2(OMe)2, P2O5, CHCl3, 80%; xv, LiOH, THF–H2O,
99%; xvi, (COCl)2, benzene; xvii, CH2N2, 63%.](/image/article/2002/CC/b111551k/b111551k-s8.gif) | ||
| Scheme 8 Reagents and conditions: i, [RhCl(COD)]2, MOD-DIOP ligand 71, H2, MeOH, 100%, er = 97∶3, ii, Br2, AcOH, 93%; iii, MsOH, P2O5, 75%; iv, H2, Pd/C, NaHCO3, MeOH, 100%; v, LiOH, THF–H2O, 100%; vi, KH, HCO2Me, DME, 0 °C, 85%; vii, methyl vinyl ketone, Et3N, CH2Cl2, 95%; viii, NaOH, H2O–THF, 95%; ix, CH2N2, Et2O–CH2Cl2, 99%; x, Br2, NaHCO3, CH2Cl2, 80%; xi, DBU, benzene, 50 °C, 90%; xii, NaBH4, i-PrOH–CH2Cl2, 99%; xiii, H2, Pd/C, MeOH, 78%, dr = 22∶1, xiv, CH2(OMe)2, P2O5, CHCl3, 80%; xv, LiOH, THF–H2O, 99%; xvi, (COCl)2, benzene; xvii, CH2N2, 63%. | ||
The product from hydrogenation of 70 was selectively brominated at C1 in order to correctly steer intramolecular Friedel-Crafts acylation of 72 towards the desired tetralone 73. Hydrogenolysis of the intervening aryl bromide and saponification was followed by a Robinson annulation of 73 with methyl vinyl ketone, and subsequent bromination yielded the dibromo phenanthrene derivative 74. Interestingly, if the carboxylic acid moiety of 73 was protected as an ester during the Robinson annulation process a degree of racemisation occurred.
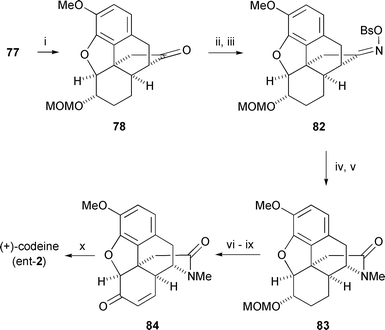
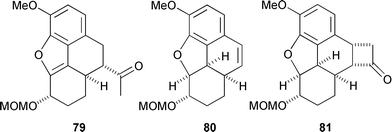
Treatment of the bromocyclohexenone 74 with 1,8-diazabicyclo[5.4.0]undec-1-ene (DBU) promoted smooth cyclisation to afford benzofuran 75via a pathway that is believed to involve isomerization of 74 to the β,γ-enone. Restoration of the original (14R) configuration in 75 is in accord with a conformational analysis which shows that a trans C9–C14 arrangement is overwhelmingly favored on thermodynamic grounds. Palladium catalysed hydrogenation of 75 resulted in an unexpected but complete reduction of the carbonyl group to a methylene unit, along with the desired reduction of the benzofuran, and it appeared at this point that we may have reached an impasse. Fortunately, metal hydride reduction of ketone 75 solved this problem by producing an equatorially oriented alcohol which was immune to hydrogenolysis. The dihydrobenzofuran 76 was formed in good yield and high stereoselectivity upon hydrogenation of this alcohol. Ester 76 was converted uneventfully into the carbenoid precursor, diazoketone 77, by four straightforward operations, setting the stage for the crucial step which connects C13 to C15. An important requirement for success in this reaction is that the ketocarbenoid at C9 and the benzylic hydrogen at C13 into which it is to insert must both be able to adopt an axial orientation, and conformational analysis convinced us that this would be the case. In the event, rhodium(II) catalysed decomposition of 77 gave the desired intramolecular C–H insertion product 78 in 50% yield (Scheme 9) along with lesser quantities of three byproducts, 79, 80 and 81. The product ratio was found to be highly dependent on the electronic and steric properties of the rhodium catalyst employed to effect decomposition, an observation that is supported by data from other laboratories.61 Formation of the benzofuran 79 is the most difficult product to rationalise via the conventional mechanism for rhodium(II) carbenoid C–H insertion and is thought to be indicative of a competing zwitterionic mechanism.62
 | ||
| Scheme 9 Reagents and conditions: i, Rh2(OAc)4, CH2Cl2, 50%; ii, HONH2·HCl, NaOAc, MeOH, 90%; iii, BsCl, DMAP, Et3N, CH2Cl2; iv, AcOH, rt, 69% (2 steps), regioselectivity = 92∶8; v, NaH, MeI, benzene, Δ 95%; vi, HBr, MeCN, 96%; vii, Dess-Martin periodinane, CHCl3, 99%; viii, t-BuOK, t-BuOH, then PhSeCl, THF, 75%; ix, NaIO4, THF–H2O, 91%; x, LiAlH4, THF, Δ 70%. | ||
The Beckmann rearrangement of the oxime derived from cyclopentanone 78 that was intended to generate the piperidine ring of morphine proved exceedingly difficult to consummate, and an alternative route from 78 via Baeyer-Villiger oxidation was entertained briefly. This tactic led to several potentially useful morphinans modified in ring D, but it failed to afford access to morphine itself.63 Fortunately, persistent experimentation was rewarded when rearrangement of oxime brosylate 82 was found to occur readily in acetic acid, the major product being the desired δ-lactam. This was N-methylated to yield tertiary amide 83 which, after a straightforward series of changes of oxidation level, afforded (+)-codeine (ent-2). Demethylation of (+)-codeine followed a protocol optimised by Rice for the natural enantiomer,64 and completed our synthesis of (+)-morphine. Although opiates of nonnatural configuration are typically devoid of analgetic activity, they often retain antitussive and other useful properties. With three routes to (+)-morphine now available in principle, it remains to be seen whether this material will garner sufficient interest to warrant further synthetic explorations in that direction.
Conclusion and outlook
The morphine story is one of the most enthralling in all of science. Its beginnings in antiquity, its evolution through the systematic efforts of Sertürner, Robinson and others, and now its prominent role as one of the trophies of contemporary synthesis, define a position for morphine that is unique among natural products. The fascination with morphine and its congeners shown by organic and medicinal chemists seems likely to continue for years to come, probably with a strong focus on those aspects of synthesis which lend practicality to the effort and which give due consideration to the impact of environmental factors in the design of any large-scale synthesis of morphine. At the time of writing this Article, the future of opium production in countries such as Afghanistan remains in doubt, and it is not inconceivable that the natural source of morphine may at some point become inaccessible or so highly controlled by hostile powers that Western medicine may be forced to find alternative means for acquiring this valuable drug. The impetus towards synthesis which this scenario provides has been noted by Hudlicky4c among others, and there is little doubt that synthetic organic chemistry will at some point in the future deliver a synthesis of morphine which is indeed ‘practical.’65,66Acknowledgements
Many graduate students and postdoctoral colleagues have been engaged in our work on morphine synthesis over the past twenty years, and credit for the modest advances we have made must go entirely to them. Their names are contained in the references. Financial support for our research in the morphine area was provided by the National Institute for Drug Abuse and the National Science Foundation.Notes and references
- Licit opium production figures were taken from the latest report of the International Narcotics Control Board (available at http://www.incb.org), and the illicit figures came from the report ‘Global Illicit Drugs Trends 2001’ published by the United Nations Office for Drug Control and Crime Prevention (available at http://www.undcp.org).
- Taken from the foreword of ref. 5a.
- (a) (a) M. Gates and G. Tschudi, J. Am. Chem. Soc., 1952, 74, 1109 CrossRef; (b) M. Gates and G. Tschudi, J. Am. Chem. Soc., 1956, 78, 1380 CrossRef CAS.
- (a) Recent reviews: C. Szántay , G. Dörnyel and G. Blaskó , in The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell and A. Brossi, Academic Press, London, 1994, 45, pp. 128–222 Search PubMed; (b) M. Maier , in Organic Synthesis Highlights II, ed. H. Waldmann, VCH, Weinheim, 1995, pp. 357–369 Search PubMed; (c) T. Hudlicky , G. Butora , S. P. Fearnley , A. G. Gum and M. R. Stabile , in Studies in Natural Products Chemistry, ed. A.-u. Rahman, Elsevier, Amsterdam, 1996, 18, pp. 43–154 Search PubMed; (d) B. H. Novak, T. Hudlicky, J. W. Reed, J. Mulzer and D. Trauner, Curr. Org. Chem., 2000, 4, 343 Search PubMed; (e) K. W. Bentley, Nat. Prod. Rep., 2000, 17, 247 RSC.
- (a) Treatises: K. W. Bentley , The Chemistry of the Morphine Alkaloids, Oxford University Press, London, 1954 Search PubMed; (b) D. Ginsburg , The Opium Alkaloids, Wiley, London, 1962 Search PubMed.
- P. T. White and S. Raymer, National Geographic, 1985, 167, 142 Search PubMed.
- A. E. Sigerest , Paracelsus in the Light of Four Hundred Years, March of Medicine, New York Academy of Medicine, New York, 1941 Search PubMed.
- G. Lindrop , The Opium-Eater. A Life of Thomas De Quincey, Dent and Sons, London, 1981 Search PubMed.
- F. W. A. Sertürner, Trommsdorff’s J. Pharm, 1806, 14, 225 Search PubMed.
- J. M. Gulland and R. Robinson, Mem. Proc. Manchester Lit. Phil. Soc., 1925, 69, 79 Search PubMed.
- A. Laurent, Ann. Chim. Phys., 1847, 19, 359 Search PubMed.
- For authoritative accounts of the structural elucidation of morphine, see: ref. 5a, chapter 1 and ref. 5b, chapter 2.
- For an excellent treatise of the opium poppy, see: L. D. Kapoor , Opium Poppy. Botany, Chemistry and Pharmacology, Haworth Press, London, 1995 Search PubMed.
- The Opiate Receptors, ed. G. W. Pasternak, Humana Press, Clifton, New Jersey, 1988 Search PubMed.
- J. Hughes, T. W. Smith, H. W. Kosterlitz, L. A. Fothergill, B. A. Morgan and H. R. Morris, Nature, 1975, 258, 577 CAS.
- The Endorphins, volume 18 in series, Advances in Biochemical Psychopharmacology, ed. E. Costa and M. Trabucchi, Raven Press, New York, 1978 Search PubMed.
- D. S. Fries , in Principles of Medicinal Chemistry, ed. W. O. Foye, T. L. Lemke and D. A. Williams, 4th Ed., Williams and Wilkins, Baltimore, 1995, Ch. 14, pp. 247–269 Search PubMed.
- O. Eislab and O. Schaumann, Dtsch. Med. Wochenschr., 1939, 65, 967.
- T. M. Kutchan , in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, London, 1998, 50, pp. 257–316 Search PubMed.
- W. De-Eknamkul and M. H. Zenk, Phytochemistry, 1992, 31, 813 CAS.
- H. Lotter, J. Gollwitzer and M. H. Zenk, Tetrahedron Lett., 1992, 33, 2443 CrossRef CAS.
- G. Mikus, A. A. Somogyi, F. Bochner and M. Eichelbaum, Biochem. Pharmacol., 1991, 41, 757 CrossRef CAS.
- D. H. R. Barton and T. Cohen , in Festschrift Arthur Stoll, Birkhauser, Basel, 1957, p. 117 Search PubMed.
- R. Pummerer, H. Puttfarcken and P. Schopflocher, Ber., 1925, 58, 1808 Search PubMed.
- D. H. R. Barton, A. M. Deflorin and O. E. Edwards, J. Chem. Soc., 1956, 530 RSC.
- D. H. R. Barton, G. W. Kirby, W. Steglich and G. M. Thomas, Proc. Chem. Soc., 1963, 203 RSC; D. H. R. Barton, Pure Appl. Chem., 1964, 9, 35 CAS.
- M. A. Schwartz and I. S. Mami, J. Am. Chem. Soc., 1975, 97, 1239 CrossRef CAS.
- M. A. Schwartz and P. T. K. Pham , J. Org. Chem., 1988, 53, 2318; and references cited Search PubMed.
- J. D. White, G. Caravatti, T. B. Kline, E. Edstrom, K. C. Rice and A. Brossi, Tetrahedron, 1983, 39, 2393 CrossRef CAS.
- C. Szántay, G. Blaskó, M. Bárczai-Beke, P. Péchy and G. Dörnyei, Tetrahedron Lett., 1980, 3509 CrossRef CAS.
- For some recent reviews of iodine(III) reagents, see: P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 Search PubMed; A. Varvoglis , Hypervalent Iodine in Organic Synthesis, Academic Press, San Diego, 1997 CrossRef CAS.
- Oxidative Coupling of Phenols, ed. A. R. Battersby and W. I. Taylor, Dekker, New York, 1967 Search PubMed.
- For some more recent applications of phenyliodoso bis(trifluoroacetate) in oxidative phenolic coupling, see: Y. Kita, M. Arisawa, M. Gyoten, M. Nakajima, R. Hamada, H. Tohma and T. Takada, J. Org. Chem., 1998, 63, 6625 Search PubMed; M. Node, S. Kodama, Y. Hamashima, T. Baba, N. Hamamichi and K. Nishide, Angew. Chem., Int. Ed., 2001, 40, 3060 CrossRef CAS.
- This useful contrivance was first introduced by Jackson and has been employed by others, see: A. H. Jackson and J. A. Martin, J. Chem. Soc. C, 1966, 2061 Search PubMed; T. Kametani, K. Shishido, E. Hayashi, C. Seino, T. Kohno, S. Shibuya and K. Fukumoto, J. Org. Chem., 1971, 36, 1295 RSC; M. A. Schwartz and M. F. Zoda, J. Org. Chem., 1981, 46, 4623 CrossRef CAS.
- J. D. White, R. J. Butlin, H.-G. Hahn and A. T. Johnson, J. Am. Chem. Soc., 1990, 112, 8595 CrossRef CAS.
- S. M. Kupchan and A. J. Liepa, J. Am. Chem. Soc., 1973, 95, 4062 CrossRef CAS.
- R. Grewe and A. Mondon, Chem. Ber., 1948, 81, 279 Search PubMed.
- R. Grewe, H. Fisher and W. Friedrichsen, Chem. Ber., 1967, 100, 1 Search PubMed.
- G. C. Morrison, R. O. Waite and J. Shavel Jr, Tetrahedron Lett., 1967, 4055 CrossRef CAS.
- T. S. Lie, L. Maat and H. C. Beyerman, Recl. Trav. Chim. Pays-Bas, 1979, 98, 419 Search PubMed.
- K. C. Rice, J. Org. Chem., 1980, 45, 3135 CrossRef CAS.
- C. Y. Hong, N. Kado and L. Overman, J. Am. Chem. Soc., 1993, 115, 11028 CrossRef CAS.
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986 CrossRef CAS.
- For the conversion of dihydrocodeinone into morphine, see: I. Iijima and K. C. Rice, Heterocycles, 1977, 6, 1157 Search PubMed.
- E. Vongerichten and H. Schrötter, Ann., 1881, 210, 396 Search PubMed.
- L. Knorr and R. Pschorr, Ber., 1889, 22, 181 Search PubMed.
- R. Pschorr, Ber., 1896, 29, 496 Search PubMed.
- R. Robinson and R. J. Ghosh, J. Chem. Soc., 1944, 506 RSC.
- D. Elad and D. Ginsburg, J. Chem. Soc., 1954, 3052 RSC.
- E. D. Bergmann, R. Pappo and D. Ginsburg, J. Chem. Soc., 1950, 1369 RSC.
- D. Ginsburg and R. Pappo, J. Chem. Soc., 1951, 516 RSC.
- D. Ginsburg and R. Pappo, J. Chem. Soc., 1951, 938 RSC.
- D. Ginsburg and R. Pappo, J. Chem. Soc., 1953, 1524 RSC.
- J. Mulzer, G. Dürner and D. Trauner, Angew. Chem., Int. Ed. Engl., 1996, 35, 2830 CrossRef CAS; J. Mulzer, J. W. Bats, B. List, T. Opatz and D. Trauner, Synlett, 1997, 441 CAS; D. Trauner, J. W. Bats, A. Werner and J. Mulzer, J. Org. Chem., 1998, 63, 5908 CrossRef CAS.
- D. Trauner, T. Opatz, S. Porth, J. W. Bats, G. Giester and J. Mulzer, Synthesis, 1998, 653 CrossRef CAS.
- K. A. Parker and D. Fokas, J. Am. Chem. Soc., 1992, 114, 9688 CrossRef CAS; K. A. Parker and D. Fokas, J. Org. Chem., 1994, 59, 3927 CrossRef CAS; K. A. Parker and D. Fokas, J. Org. Chem., 1994, 59, 3933 CrossRef CAS.
- J. F. Ruppert and J. D. White, Chem. Commun., 1976, 976 RSC.
- D. F. Taber , in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, 3, pp. 1045–1062 Search PubMed.
- J. D. White, P. Hrnciar and F. Stappenbeck, J. Org. Chem., 1997, 62, 5250 CrossRef CAS; J. D. White, P. Hrnciar and F. Stappenbeck, J. Org. Chem., 1999, 64, 7871 CrossRef CAS.
- T. Morimoto , M. Chiba and K. Achiwa , Chem. Pharm. Bull, Japan, 1993, 41, 1149 Search PubMed.
- M. P. Doyle, V. Bagheri, T. J. Wandless, N. K. Harn, D. A. Brinker, C. T. Eagle and K.-L. Loh, J. Am. Chem. Soc., 1990, 112, 1906 CrossRef CAS.
- J. D. White and P. Hrnciar, J. Org. Chem., 1999, 64, 7271 CrossRef.
- J. D. White and P. Hrnciar, J. Org. Chem., 2000, 65, 2646 CrossRef CAS.
- K. C. Rice, J. Med. Chem., 1977, 20, 164 CrossRef CAS.
- For a practical synthesis of (+)-morphine, (+)-codeine and (+)-heroin from a related alkaloid, see: I. Iigima, J.-i. Minamikawa, A. E. Jacobson, A. Brossi and K. C. Rice, J. Org. Chem., 1978, 43, 1462 Search PubMed.
- A new asymmetric synthesis of (−)-morphine has been reported very recently: see H. Nagata, N. Miyazawa and K. Ogasawara, Chem. Commun., 2001, 1094 Search PubMed.
Footnotes |
| † 54% illicit, enough to manufacture 470 tons of heroin. |
| ‡ 71% illicit, mainly in Myanmar (Burma) and Afghanistan. |
| § India is currently the world’s largest legal producer of opiate raw materials, closely followed by Australia. |
| This journal is © The Royal Society of Chemistry 2002 |