Synthesis, crystal and molecular structure of [Sn(η4-P2C2But2)]: the first non transition metal 1,3-diphosphacyclobutadienyl compound
Matthew D.
Francis
* and
Peter B.
Hitchcock
School of Chemistry, Physics and Environmental Science, University of Sussex, Brighton, UK BN1 9QJ. E-mail: m.d.francis@sussex.ac.uk
First published on 21st December 2001
Abstract
Treatment of [Zr(η5-C5H5)2(PCBut)2] with SnCl2 led to the novel monomeric (1,3-diphosphacyclobutadienyl)tin(II) half sandwich complex [Sn(η4-P2C2But2)] which has been characterised by multinuclear NMR spectroscopy and in the solid state by a single crystal X-ray diffraction study.
In 1986 the groups of Nixon and Binger independently reported the first examples of phosphaalkyne (P
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR) cyclodimerisation in the coordination sphere of cobalt, rhodium and iridium, yielding complexes of the previously unknown 1,3-diphosphycycloabutadiene ring.1 Subsequently, further examples appeared containing zero-valent nickel, iron and molybdenum.2 Such compounds have generally been prepared via displacement, by phosphaalkynes, of a labile transition metal bound substituent (e.g. ethylene) followed by a [2 + 2] cycloaddition or by co-condensation of metal atoms with phosphaalkynes by the metal vapour synthesis technique (MVS). In contrast, main group compounds of the 1,3-diphosphacyclobutadiene ring have been unknown until now. Furthermore, it could be anticipated that such compounds would not be readily accessible via straightforward cyclodimerisation of phosphaalkynes. Recently, however,
we have succeeded in the preparation of the first main group diphosphacyclobutadiene complex namely [Sn(η4-P2C2But2)] 1. Herein, we report the synthesis of 1 from the reaction between SnCl2 and the 1,3-diphosphabicyclo[1.1.0]butane zirconium complex [Zr(η5-C5H5)2(PCBut)2]32.
CR) cyclodimerisation in the coordination sphere of cobalt, rhodium and iridium, yielding complexes of the previously unknown 1,3-diphosphycycloabutadiene ring.1 Subsequently, further examples appeared containing zero-valent nickel, iron and molybdenum.2 Such compounds have generally been prepared via displacement, by phosphaalkynes, of a labile transition metal bound substituent (e.g. ethylene) followed by a [2 + 2] cycloaddition or by co-condensation of metal atoms with phosphaalkynes by the metal vapour synthesis technique (MVS). In contrast, main group compounds of the 1,3-diphosphacyclobutadiene ring have been unknown until now. Furthermore, it could be anticipated that such compounds would not be readily accessible via straightforward cyclodimerisation of phosphaalkynes. Recently, however,
we have succeeded in the preparation of the first main group diphosphacyclobutadiene complex namely [Sn(η4-P2C2But2)] 1. Herein, we report the synthesis of 1 from the reaction between SnCl2 and the 1,3-diphosphabicyclo[1.1.0]butane zirconium complex [Zr(η5-C5H5)2(PCBut)2]32.

Treatment of 2 with one equivalent of SnCl2 in thf at 60 °C or in toluene at reflux led to the formation of the novel half sandwich tin(II) compound [Sn(η4-P2C2But2)] 1 (Scheme 1). Extraction with hexane of the crude reaction mixture followed by filtration and evaporation of the solvent afforded 1 as a bright yellow powder in 86% yield. This material was analytically pure and needed no further purification.† Compound 1 is remarkably stable. It melts without decomposition at 154–156 °C and the 31P{1H} NMR spectrum shows no detectable decomposition even after the solution has been exposed to air for 24 h. Spectroscopic data for 1† are consistent with its proposed half sandwich structure which was subsequently confirmed by a single crystal X-ray diffraction study (vide infra). The EI mass spectrum of 1 shows a strong molecular ion with the expected isotopic pattern. The 31P {1H} NMR spectrum of 1 consists of a single resonance at 144.9 ppm flanked by clearly defined tin satellites which indicate 1JP–Sn(119) and 1JP–Sn(117) couplings of 296.1 and 283.6 Hz, respectively. This chemical shift is notably higher than that seen in transition metal complexes of the 1,3-diphosphacyclobutadiene ring e.g. 39 ppm in [Co(η5-C5Me5)(η4-P2C2But2)]13 and 81.2 ppm in [Ni(η4-P2C2But2)2]24. The coupling of 296.1 Hz is reflected in the 119Sn{1H} NMR spectrum which shows a sharp triplet at −2128.9 ppm. Interestingly, this chemical shift is very close to those seen for a range of cyclopentadienyl tin(II) derivatives e.g. −2171.1 in [Sn(η5-C5H4Me)2]4 and −2129 in [Sn(η5-C5Me5)2].5 These chemical shifts, which are some 2000 ppm to high field of their Sn(IV) analogues, have been attributed to high coordination number and concomitant high shielding.6 Initially, the 13C NMR spectrum of 1 was puzzling with no evidence of either the ring or But quaternary signals. A refocused INEPT experiment at higher operating frequency, however, revealed both signals which had been previously obscured by the solvent and methyl carbon signals. Furthermore, it was possible to measure the Sn couplings to both quaternary signals.†
 | ||
| Scheme 1 Reagents and conditions: i, SnCl2, thf 60 °C or PhMe 110 °C. | ||
The molecular structure of 1‡ is depicted in Fig. 1. Each molecule lies on a twofold rotation axis. The geometry of the 1,3-diphosphacyclobutadiene ring is comparable with that seen in a number of transition metal complexes bearing this ligand. The phosphorus–carbon bond lengths of 1.796(3) and 1.800(3) Å are essentially equivalent and very close to those in [Co(η5-C5Me5)(η4-P2C2But2)] 3 (1.79(1)–1.82(1) Å) and [Ni(η4-P2C2But2)2] 4 (1.793(9)–1.806(8) Å). Likewise, the P–C–P and C–P–C angles in 1 of 97.47(17) and 82.11(17)°, respectively, are in close agreement with those of 98.0(5) and 82.0(5)° in 3 and 97.9(4) and 81.3(4)° in 4.
 | ||
| Fig. 1 Molecular structure of 1. Selected bond lengths (Å) and angles (°): Sn–Cl 2.432(3), Sn–P 2.6109(10), P–Cl 1.796(3), P–Cl′ 1.800(3), Sn–Ct 2.180 (Ct = centroid of P2C2 ring); Cl–P–Cl′ 82.11(17), P–Cl–P′ 97.47(17). | ||
The P2C2 ring in 1 deviates very slightly from planarity, the dihedral angle between the planes defined by P–Cl–P′ and P–Cl′–P′ being 5.9(1)°. A similar but even more pronounced folding of the rings has been seen in the zero-valent molybdenum complex [Mo(η4-P2C2But2)3] 5 in which the fold angles of the three individual rings are 10.66, 19.91 and 25.34°.
Although the molecules are stacked in linear chains in the crystal (Fig. 2), there appear to be no significant intermolecular interactions. The distance between the tin atom and the centroid of nearest non-bonded ring is 3.906 Å which is almost twice the distance of 2.180 Å between the tin and the centroid of the ring within a molecule.
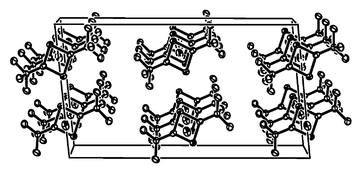 | ||
| Fig. 2 Unit cell diagram of 1. | ||
With regards to the mechanism of formation of 1, it seems likely that zirconocene dichloride is eliminated with formation of an initial intermediate 6 which subsequently rearranges to 1. It has already been shown that suitable main group halides can displace zirconocene dichloride from 2 with the formation of stable compounds similar in structure to 6, e.g.7 and 8.7 Given that 2 as well as the possible intermediate 6 both contain a P–P bond it may seem surprising that the isomeric (1,2-diphosphacyclobutadienyl)tin(II) analogue is not observed. Examples of complexes containing the 1,2-diphosphacyclobutadiene ring e.g. [Ti(η4-P2C2But2)(η8-C8H8)]8 are however rare. This is probably partly due to the unfavourable steric repulsion between the bulky But groups since head-to-head cyclodimerisation of phosphaethyne, at least under thermal conditions, has been calculated to be more favourable than head-to-tail cyclodimerisation.9 In terms of bonding, the 1,3-diphosphacyclobutadiene ring is usually though of as a neutral 4π electron donor as in the formally zero-valent transition metal complexes 4 and 5. In the case of 1, however, in which the tin centre is undoubtedly Sn(II), the ligand is probably best thought of as accepting two electrons from the metal centre thus existing as its aromatic 6π electron dianion. A theoretical study of 1 as well as a study of its reactivity is in progress, the results of which will be reported in a furture publication.

The Leverhulme Trust is gratefully acknowledged for supporting this work through a Special Research Fellowship (to M. D. F.). Dr Anthony Avent is thanked for assistance with NMR measurements.
Notes and references
- K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy, Wiley, Chichester, 1998, p. 258 et seq.; Search PubMed; P. B. Hichcock, M. J. Maah and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1986, 737 Search PubMed; P. B. Binger, R. Milczarek, R. Mynott, M. Regitz and W. Rösch, Angew. Chem., Int. Ed. Engl., 1986, 25, 644 RSC; P. Binger, R. Milczarek, R. Mynott, C. Krüger, Y. H. Tsay, E. Raabe and M. Regitz, Chem. Ber., 1988, 121, 637 CrossRef.
- M. Dreiss, D. Hu, H. Pritzkow, H. Schäufele, U. Zenneck, M. Regitz and W. Rösch, J. Organomet. Chem., 1987, 334, C35 CrossRef; T. Wettling, G. Wolmershäuser, P. Binger and M. Regitz, J. Chem. Soc., Chem. Commun., 1990, 1541 RSC; F. G. N. Cloke, K. R. Flower, P. B. Hitchcock and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1994, 489 RSC.
- T. Wettling, B. Geissler, R. Schneider, S. Barth, P. Binger and M. Regitz, Angew. Chem., Int. Ed. Engl., 1992, 31, 758 CrossRef.
- A. Bonny, A. D. McMaster and S. R. Stobart, Inorg. Chem., 1978, 17, 935 CrossRef CAS.
- B. Wrackmeyer, E. Kupce, G. Kehr and A. Sebald, Magn. Reson. Chem., 1992, 30, 964 CAS.
- B. Wrackmeyer, Annu. Rep. NMR Spectrosc., 1999, 38, 203 Search PubMed.
- P. Binger, T. Wettling, R. Schneider, F. Zurmühlen, U. Bergsträsser, J. Hoffmann, G. Maas and M. Regitz, Angew. Chem., Int. Ed. Engl., 1991, 30, 207 CrossRef.
- P. Binger, G. Glaser, S. Albus and C. Krüger, Chem. Ber., 1995, 128, 1261 CrossRef CAS.
- M. T. Nguyen, L. Landuyt and L. G. Vanquickenborne, J. Org. Chem., 1993, 58, 2817 CrossRef CAS.
- G. M. Sheldrick, SHELX-97 suite of programs for crystal structure analysis, University of Göttingen, Göttingen, Germany, 1997..
Footnotes |
| † Synthesis, spectroscopic and analytical data for 1: SnCl2 (470 mg, 2.48 mmol) and 2 (950 mg, 2.26 mmol) were placed in an ampoule and thf (30 mL) was added. The resulting mixture was stirred at 60 °C for 48 h. Volatiles were removed from the resulting orange–brown solution and the residue was extracted with hexane (2 × 30 mL) and filtered to give a yellow solution. The hexane was removed in vacuo to afford 1 as a bright yellow, pure crystalline solid (0.620 g, 86%).NMR (C6D6, 298 K): δH (300 MHz) 1.03 (s, 18H, But). δP (121.68 MHz) 144.9 (s, 1JP–Sn(119) 296.1, 1JP–Sn(117) 283.6 Hz, from satellites). δC (125.72 MHz) 34.2 (t, 3JPC 4.14 Hz, –C(CH3)3), 34.0 (t, 2JPC 6.29, 2JSnC 9.26 Hz, –C(CH3)3), 129.0 (t, JPC 48.86, 1JC–Sn(119) 110.9, 1JC–Sn(117) 105.9 Hz, PCP), δSn (186.36 MHz) −2128.9 (t, 1JPSn 296.1 Hz). EI mass spectrum (main peaks) (70 eV) m/z (%): 318 (65) M+, 303 (66) [M − CH3]+, 119 (23) Sn+. Microanalysis. Found: C 37.18; H, 5.62. C10H18P2Sn requires C, 37.67; H, 5.69%; mp 154–156 °C. |
| ‡ Crystal data for 1: single crystals were grown from a slowly cooled toluene solution. C10H18P2Sn, M = 318.87, monoclinic, space group C2/c (no. 15), a = 19.812(2), b = 6.0863(8), c = 10.6752(13) Å, β = 96.024(7)°, V = 1280.1(3) Å3, Z = 4, μ = 2.21 mm−1, 2518 reflections collected, 1115 independent (Rint = 0.057), final R indices: R1 = 0.034, wR2 = 0.083 for 998 reflections with I > 2σI; R1 = 0.039, wR2 = 0.086 for all data.Intensity data were measured on a KappaCCD area detector at 173(2) K using Mo-Kα radiation (λ = 0.71073 Å). The structure was solved by direct methods and refined on F2 using full matrix least squares with SHELX-97.10 An empirical absorption correction was applied.CCDC reference number 174129.See http://www.rsc.org/suppdata/cc/b1/b110228c/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2002 |