First example of a covalently bound dimeric inverted porphyrin†
Izabela
Schmidt
and
Piotr J.
Chmielewski
*
Department of Chemistry, University of Wroclaw, F. Joliot-Curie 14, Wroclaw 50 383, Poland. E-mail: pjc@wchuwr.chem.uni.wroc.pl; Fax: (48 071) 32 823 48; Tel: (48 071) 37 57 277
First published on 11th December 2001
Abstract
Reaction of 5,10,15,20-tetraphenyl-2-aza-21-carbaporphyrinatonickel(II) with dihalomethanes in the presence of a proton scavenger gives a 2,21′-CH2-linked dimer of Ni(II) inverted porphyrins with the yield reaching 90% and its 2,2′-linked isomer as a minor product.
The chemistry of porphyrin analogs, homologues, and isomers is a broad and exciting field.1 Among porphyrin isomers are the so-called inverted or N-confused porphyrins2 and doubly N-confused porphyrin3 which possesses a carbon atom(s) directed toward the macrocyclic crevice and nitrogen atom(s) pointing outwards. Discovery of the meso-tetraphenyl inverted porphyrin 1 among the products of the Rothemund synthesis and subsequent optimization of its yield4 make this macrocycle a readily obtainable and therefore attractive subject of studies and modifications. In addition to porphyrin-like properties, the inverted porphyrin offers unique possibilities related to the reactivity of the inverted pyrrole. Each of its three unsubstituted positions i.e. the external nitrogen,5 the adjacent CH unit,6 and internal carbon,7,8 may be a target of a substitution. Particularly interesting is the reactivity of organometallic transition metal complexes of inverted porphyrin such as 2 and its homologue 3 towards alkylation8,9 that involves internal carbon. Methylation of 2 leads to a complex 4 that possesses an sp3 carbon atom coordinated to a low-spin nickel(II) ion.8

Here we report on a reaction of the nickel(II) complex of inverted porphyrin with dihalomethanes resulting in the bis-nickel(II) complexes of the first covalently bound inverted porphyrin dimers, namely 2-(21′-(5′,10′,15′,20′-tetraphenyl-2′- aza-21′-carbaporphyrinatonickel(II)))-methyl-5,10,15,20-tetraphenyl-2-aza-21-carbaporphyrinatonickel(II) 5 and isomeric bis-(2,2′-(5,10,15,20-tetraphenyl-2-aza-21-carbaporphyrinatonickel(II))methane 6 as a minor product (Scheme 1).
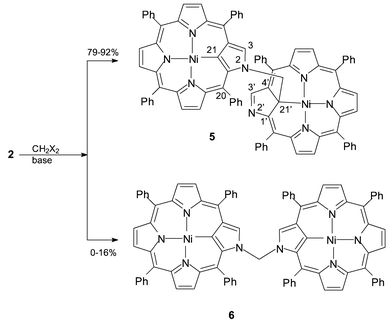 | ||
| Scheme 1 | ||
5,10,15,20-Tetraphenyl-2-aza-21-carbaporphyrinatonickel(II) 2 slowly reacts at room temperature in a CH2Cl2 solution with an excess of CH2Br2 or CH2I2 in the presence of proton scavenger.‡ For CH2Br2 the molar ratio of 5 to 6 in the reaction mixture increases from about 5∶1 to more than 10∶1 when the process is carried out under reflux for 5 h while for methylene iodide only 5 is formed with the yield exceeding 90%.
Mass spectrometry confirms the presence of two porphyrin units in 5 and 6 while 1H and 13C NMR data afford definitive structural information. Analysis of the respective chemical shifts, scalar 1H–1H and multibond 1H–13C couplings as well as 1H–1H through-space contacts allows the construction of a reliable molecular model11 (see the supplementary data).†
Proton NMR spectrum of 5 (Fig. 1) consists of fourteen pyrrolic signals unequivocally indicating the asymmetric character of the dimer. The low-field signals (3′, p′) are attributable to the pyrrole protons of the 21′-substituted fragment of the molecule.8 The signals of β-pyrrole protons of the 2-substituted part (p in Fig. 1) are spread over the region from 8.2 to 6.9 ppm. The most diagnostic spectral features of 5 are the signals of the ‘inverted pyrrole’ protons (3 and 3′) of both molecule fragments and the high-field shifted diastereotopic pair of protons of the methylene linker (2,21′-CH2).
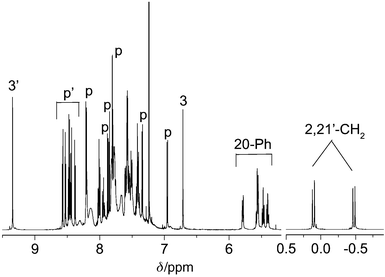 | ||
| Fig. 1 500 MHz 1H NMR spectrum of 5, CDCl3, 298 K. Assignments: see text and Scheme 1. | ||
The 1H NMR spectrum of 6 shows only one set of seven pyrrole signals indicating effective two-fold symmetry of the molecule. The signal of the methylene linker is a singlet (5.55 ppm, CDCl3, 213 K) and gives a NOESY correlation with the signal of proton 3 (7.94 ppm).
The selectivity of the reaction is noteworthy. It should be pointed out that at least five different products of the reaction are possible including 2- or 21-halomethane-substituted monomers, two ‘symmetrical’ dimers, i.e. those with a 2,2′-CH2- or 21,21′-CH2-link, and an ‘unsymmetrical’ dimer with a 2,21′-CH2-bridge that is overwhelmingly the preferred product.
Although an excess of applied dihalomethane should disfavor dimers, no monomeric halomethane-substituted products have been isolated or observed in the reaction mixture neither by mass spectrometry nor by NMR even when the reaction is quenched before completion. Thus the reaction mechanism presumably involves slow initial formation of halomethane-substituted monomer followed by its immediate consumption by an unsubstituted molecule of 2. It implies activation of the X–CH2 bond in the monomer and/or permanent excess of the unsubstituted 2 over the halomethane-substituted intermediate in the reaction mixture. One can anticipate that the inner carbon 21-C is a primary alkylation target as it is in the case of methylation of 2 by methyl iodide leading to 4.8 It is also in line with the distribution of the reaction products: the 2,2′-linked dimer 6 is only a minor product while formation of 5 dominates. Formation of a 21,21′-CH2-linked dimer may be excluded by steric factors if the associative mechanism is operative for the reaction step involving linking of porphyrinoids.
Owing to its selectivity the reaction is a convenient way to diporphyrinic systems containing two inequivalent coordination sites that are relatively close to each other. Moreover, the unsubstituted nitrogen 2′ may serve as an external protonation/coordination site or hydrogen bond acceptor making 5 a potential building block of molecular assemblies.
The reversible protonation of 2′-N changes the spin state of nickel(II) in 21′-substituted fragment from singlet to triplet due to concurrent ligation of an anion to the nickel ion analogously as in the case of 4.8 Site-specific deuteration of all pyrrole position (5[D14]-HCl), application of deuterated acid, and COSY experiments allow partial assignment of paramagnetically shifted protons in the 1H NMR spectrum of 5-HCl (Fig. 2, CDCl3, 298 K). Among the seven low-field shifted pyrrole signals there is one (20.9 ppm) that belongs to the 2-substituted part of the molecule. It can be assigned to the proton 3 that is located at the shortest distance to the paramagnetic center. The remained signals appear in the much narrower, crowded region of −4–12 ppm; their chemical shifts, however, are temperature dependent. Thus, although the nickel(II) ion in the 2-substituted subunit remains low-spin, there is a spin-density transfer on this part of the molecule.
![(A) 500 MHz 1H NMR spectrum of 5-HCl in CDCl3, 298 K; scalar couplings of β-pyrrole protons are marked. (B) 76 MHz 2H NMR of 5[D14]-HCl in CHCl3, 298 K.](/image/article/2002/CC/b109760c/b109760c-f2.gif) | ||
| Fig. 2 (A) 500 MHz 1H NMR spectrum of 5-HCl in CDCl3, 298 K; scalar couplings of β-pyrrole protons are marked. (B) 76 MHz 2H NMR of 5[D14]-HCl in CHCl3, 298 K. | ||
Cyclic voltammetry of 5 (CH2Cl2, TBAP, glassy carbon electrode) shows two one-electron quasi-reversible (678 and 810 mV vs. SCE) and one irreversible (anodic peak at 1110 mV) oxidation processes. E1/2 of the least anodic couple and the potential of the irreversible oxidation are similar to those observed for 2 or 3.10 On the other hand no couple corresponding to that at 810 mV occurs for monomeric nickel(II) complexes of inverted tetraaryl porphyrins or its homologues. In particular, it does not resemble that of the structurally related monomeric reference complex 4 for which there is an irreversible oxidation observed with the anodic peak at 671 mV and the cathodic one at 503 mV. It seems that the oxidation of one fragment of the system strongly modifies redox properties of the other electroactive part of 5 indicating interaction between the redox centers of the dimer.12
In conclusion, we have shown that the formation of asymmetric, 2,21′-CH2-linked nickel(II) complex of tetraphenyl inverted diporphyrin is highly selective. To some extent 5 can be described as a superposition of 2- and 21-alkylated inverted porphyrin nickel(II) complexes represented here by the reference compounds 3 and 4. Although aromatic systems of the porphyrinic subunits in 5 are not coupled, interaction between them can be observed.
This work was supported by the State Committee for Scientific Research of Poland (Grant 7 T09A 120 20 and 3 09A 155 15).
Notes and references
- J. L. Sessler, A. Gebauer, E. Vogel, in The Porphyrin Handbook, vol. 2, ch. 8; Search PubMed; L. Latos-Grazynski, in The Porphyrin Handbook, ed. K. K. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 1999, vol. 2, ch. 14. Search PubMed.
- P. J. Chmielewski, L. Latos-Grazynski, K. Rachlewicz and T. Glowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779 CrossRef; H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767 CrossRef CAS; B. Y. Liu, C. Brückner and D. Dolphin, Chem. Commun., 1996, 2141 RSC.
- H. Furuta, H. Maeda and A. Osuka, J. Am. Chem. Soc., 2000, 122, 803 CrossRef CAS.
- G. R. Geier III, D. M. Haynes and J. S. Lindsey, Org. Lett., 1999, 1, 1455 CrossRef.
- P. J. Chmielewski and L. Latos-Grazynski, J. Chem. Soc., Perkin Trans. 2, 1995, 503 RSC.
- I. Schmidt and P. J. Chmielewski, Tetrahedron Lett., 2001, 42, 1151 CrossRef CAS.
- Y. Ishikawa, I. Yoshida, K. Akaiwa, E. Koguchi, T. Sasaki and H. Furuta, Chem. Lett., 1997, 453 CrossRef CAS.
- P. J. Chmielewski, L. Latos-Grazynski and T. Glowiak, J. Am. Chem. Soc., 1996, 118, 5690 CrossRef CAS.
- P. J. Chmielewski, L. Latos-Grazynski and I. Schmidt, Inorg. Chem., 2000, 39, 5475 CrossRef CAS.
- P. J. Chmielewski and L. Latos-Grazynski, Inorg. Chem., 1997, 36, 840 CrossRef CAS.
- HyperChem Release 5.01 MM+ forcefield optimized by Polak-Ribiere.
- J. Y. Becker, D. Dolphin, J. B. Paine and T. Wijesekera, J. Electroanal. Chem., 1984, 164, 335 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, NOESY and HMBC data and molecular models for 5 and 6, NMR data for 5-HCl, cyclic voltammograms for 3–5 and electronic spectra for 5 and 6. See http://www.rsc.org/suppdata/cc/b1/b109760c/ |
| ‡ In a typical synthesis 30 mg of 2 (0.045 mmol) was mixed with methylene bromide (1.42 mmol) in deaerated dichloromethane (30 cm3) in the presence of 10 mg of solid potassium carbonate and 2 mg of dibenzo-18-crown-6 as a phase transfer agent. The inert atmosphere was kept to prevent the nickel center from autooxidation under basic conditions.10 The 1H NMR signals of the starting complex 2 disappeared after three days of stirring at rt. Flash chromatography on the silica gel with CH2Cl2 allows the separation of reaction products and the crown ether. Yields (crystallized from CH2Cl2–hexane): 5, 24 mg, (79%); 6, 5 mg, (16.5%). Selected data for 5: λmax(CH2Cl2)/nm (log ε) 242 sh, 284 (4.64), 357 (4.76), 431 (5.06), 465 sh, 564 (4.29), 718 (3.76), 788 (3.70); MS (ESI) calc. for C89H56N8Ni2 + H: 1355.9. Found: 1355.4. Calc. for C89H56N8Ni2·2H2O: C, 76.86; H, 4.35; N, 8.06. Found: C, 76.58; H, 4.33; N, 8.13%. Selected data for 6: λmax(CH2Cl2)/nm (log ε) 360 (4.76), 429 (4.92), 468 (4.79) 566 (4.01), 611 sh, 726 (3.74), 801 (3.70); MS (ESI) calc. for C89H56N8Ni2 + H: 1355.9. Found: 1355.3. |
| This journal is © The Royal Society of Chemistry 2002 |