Enhancing the reactivity of uranium(VI) organoimido complexes with diazoalkanes†
Jaqueline L.
Kiplinger
*,
David E.
Morris
,
Brian L.
Scott
and
Carol J.
Burns
*
Chemistry Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.. E-mail: cjb@lanl.gov
First published on 3rd December 2001
Abstract
Diphenyldiazomethane effects a two-electron oxidation of the uranium(IV) monoimido complex (C5Me5)2U(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-2,4,6-t-Bu3C6H2) to give the uranium(VI) mixed bis(imido) complex, (C5Me5)2U(N-2,4,6-t-Bu3C6H2)(N–NCPh2), which undergoes a rare cyclometallation reaction upon mild thermolysis to afford a uranium(IV) bis(amide) complex that results from net addition of a C–H bond of an ortho tert-butyl group across the NUN core.
N-2,4,6-t-Bu3C6H2) to give the uranium(VI) mixed bis(imido) complex, (C5Me5)2U(N-2,4,6-t-Bu3C6H2)(N–NCPh2), which undergoes a rare cyclometallation reaction upon mild thermolysis to afford a uranium(IV) bis(amide) complex that results from net addition of a C–H bond of an ortho tert-butyl group across the NUN core.
Arguably, one of the most exciting discoveries over the past 15 years in organometallic chemistry has been the observation that high-valent (d0) early transition-metal imido complexes readily activate carbon–hydrogen bonds.1 In addition, these compounds have been shown to display a rich chemistry1,2 that includes cycloadditions of unsaturated C–C and C–X bonds, addition of H2 and alkylsilanes, hydroamination of alkynes to enamines, and recently, catalytic imine metathesis.3 High-valent (f0) actinide imido complexes might be expected to display similar reactivity patterns to their early transition-metal counterparts; however, the most characteristic chemical property of uranium imido (U
N) bonds is their decided lack of reactivity. Presumably, this is a consequence of diminished polarity of the uranium–nitrogen bond due to higher
bond order resulting from nitrogen 2p lone-pair donation to uranium, higher available coordination numbers for uranium, and the ability of uranium to access 5f-orbitals for multiple bonding.4
The reaction chemistry of uranium(VI) bis(imido) complexes is limited to reduction by H24 and arylsilanes5 to form the corresponding U(IV) bis(amide) complexes, catalytic disproportionation of 1,2-diphenylhydrazine to afford aniline and azobenzene,6 and a recent report involving the intramolecular activation of a pentamethylcyclopentadienyl methyl C–H bond across the two imido functional groups of a U(VI) bis(imido) complex to give the corresponding U(IV) bis(amide) ‘tuck-in’ complex.7 Importantly, most of these reactions were promoted under forcing conditions (elevated temperatures).
In our ongoing pursuit to develop synthetic entries towards uranium complexes containing multiply bonded functional groups, we discovered a method to access reactive uranium organoimido complexes. We now report that diazoalkanes can be used to prepare uranium(VI) bis(imido) complexes which are reactive toward sp3 hybridized C–H bonds. This chemistry represents a new mode of intramolecular C–H activation for organoactinide complexes.8 The key feature of this work is that net 1,3 addition of a C–H bond across the NUN core of an uranium(VI) mixed-bis(imido) complex occurs under mild conditions to afford the corresponding bis(amide) complex.
As depicted in Scheme 1, treatment of a toluene solution of the uranium(IV) monoimido complex 1 with an equimolar amount of diphenyldiazomethane generates the uranium(VI) bis(imido) complex 2 as a brown crystalline solid in 97% yield.9
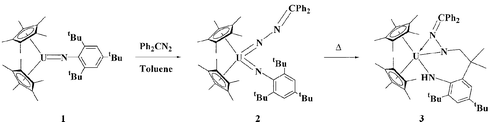 | ||
| Scheme 1 | ||
Complex 2 does not lose N2 to give the uranium(VI) alkylidene complex. This in marked contrast with the chemistry observed for isoelectronic N2O and organoazides (N3R) which have been exploited as valuable two electron oxidative transfer agents to uranium(IV) imido precursors for the preparation of uranium(VI) oxo and uranium(VI) imido complexes, respectively.4 It is important to note that although the reaction between a transition metal complex and a substituted diazomethane to produce a complex that contains a metal–nitrogen multiple bond is a reaction type that has been known for the past 20 years,10 this work represents the first time that diazoalkanes have been utilized as two-electron oxidants at an actinide metal center.
That the uranium metal center has been oxidized from U(IV) to U(VI) is clearly demonstrated by the 1H NMR and electronic absorption spectra of 2. The room-temperature electronic absorption spectrum, recorded in toluene solution from 1600 to 300 nm, shows no f→f transitions in the near IR region, but does show a broad, intense, and featureless charge-transfer band in the visible region, which is consistent with the assignment of an f0 U(VI) metal center.11 Also, variable-temperature 1H NMR spectroscopy (−75 to +60 °C) reveals that the 1H NMR spectrum of 2 is temperature invariant and indicates that complex 2 behaves like a temperature-independent paramagnet (TIP), a characteristic property of U(VI) in related organometallic species.11
The identity of complex 2 as a uranium(VI) bis(imido) complex was confirmed by a single-crystal X-ray diffraction study (Fig. 1(a)).‡ The molecular structure of 2 reveals a typical bent metallocene framework
with a pseudotetrahedral coordination environment about the uranium atom; the two imido ligands are terminally bound to the uranium metal center. The uranium–nitrogen bond lengths (U(1)–N(3) 1.987(5) Å and U(1)–N(1) 2.031(6) Å) and nearly linear U–N–Cipso and U–N–N bond angles (U(1)–N(3)–C(34) 176.1(5)° and U(1)–N(1)–N(2) 157.1(5)°) are consistent with the assignment of the ligands as organoimido groups. The uranium–nitrogen bond distances lie at the high end of the range for other structurally characterized uranium(VI) complexes containing terminal imido groups (e.g. (C5Me5)2U(NPh)2: U–N 1.952(7) Å;11b (C5Me5)2U(NAd)2: U–N 1.94(2), 1.96(2) Å;12 (C5Me5)2U(O)(N-2,6-i-Pr2C6H3):
U–N 1.988(4) Å;11c [N(SiMe3)2]3U(F)(NPh): U–N 1.979(8) Å).11a The lengthening of the UN bonds is most likely a manifestation of the immense steric demands dictated by the 2,4,6-t-Bu3C6H2 group. Importantly, the metrical parameters associated with the fragment derived from the diazoalkane supports the formation of an uranium–imido linkage; the bound diazoalkane has been reduced as evidenced by the significant elongation of the N(1)–N(2) bond distance (1.308(8) Å) compared with that of uncomplexed diazoalkanes (1.12–1.13 Å).13
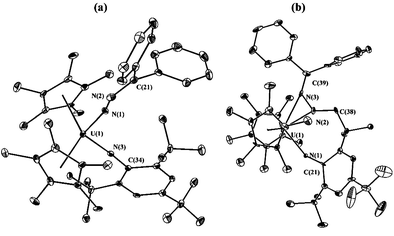 | ||
| Fig. 1 (a) Molecular structure of 2 with thermal ellipsoids at the 20% probability level. Selected bond distances (Å) and angles (°): U(1)–N(1) 2.031(6), U(1)–N(3) 1.987(5), N(1)–N(2) 1.308(8), N(2)–C(21) 1.310(10); N(1)–U(1)–N(3) 104.9, U(1)–N(1)–N(2) 157.1(5), N(1)–N(2)–C(21) 130.3(7), U(1)–N(3)–C(34) 176.1(5). (b) Molecular structure of 3 with thermal ellipsoids at the 20% probability level. Selected bond distances (Å) and angles (°): U(1)–N(1) 2.315(9), U(1)–N(2) 2.228(10), U(1)–N(3) 2.496(12), N(2)–N(3) 1.364(13), N(1)–C(21) 1.404(14), N(2)–C(38) 1.461(14), N(3)–C(39) 1.325(16); N(1)–U(1)–N(2) 81.9(4), N(2)–U(1)–N(3) 32.9(3), U(1)–N(1)–C(21) 133.2(8), U(1)–N(2)–C(38) 150.2(9), U(1)–N(3)–C(39) 168.3(9). | ||
Mild thermolysis of a toluene-d8 solution of 2 (100 °C, 20 min) results in a marked color change from brown to dark cherry red and 1H NMR spectroscopy signals the quantitative formation of the novel cyclometallated uranium(IV) bis(amide) complex 3 (Scheme 1); the NMR spectrum of the product is paramagnetically shifted, which indicates that the product is a reduced U(IV) species. Following work-up, the isolated yield for complex 3 is 73%.9
That the C–H bond of one of the ortho tert-butyl groups from the UN-2,4,6-t-Bu3C6H2 fragment has been activated is unambiguously ascertained by X-ray crystallography which clearly shows the net 1,3 addition of the C–H fragment across the NUN core (Fig. 1(b)).‡ As with the bis(imido) complex 2, complex 3 possesses the standard pseudotetrahedral geometry observed for bent metallocene uranium complexes. In the metallocene wedge lie three nitrogen atoms: two are associated with uranium–amide linkages (U(1)–N(1) 2.315(9) Å and U(1)–N(2) 2.228(10) Å) and one is an uranium–nitrogen dative interaction (U(1)–N(3) 2.496(12) Å). Notably, the U(1)–N(1)–C(21) bond angle is 133.2(8)° which deviates significantly from the nearly linear U–N–Cipso
angle (176.1(5)°) present in the bis(imido) complex 2.
Perhaps the most striking aspect of the cyclometallation sequence is the net 1,3 addition of the C–H bond from one of the ortho tert-butyl groups across the NUN fragment; this reaction type has no equivalent in the transition metal series and represents a new mode of C–H activation available for imido ligands.7,8 The mechanism for this unusual transformation is not established yet. However, two mechanistic scenarios are proposed to rationalize the observed chemistry. One explanation invokes a stepwise process with initial addition of the C–H bond across one UN fragment to generate the U(VI) hydride intermediate (A) which undergoes a 1,2-hydride shift to give the observed U(IV) bis(amide) complex (3). An alternative route is a thermally allowed (2π + 2π + 2σ) pericyclic reaction in which the C–H bond is transferred across the NUN fragment
in a concerted fashion (B). The mechanism of this reaction is currently under investigation.
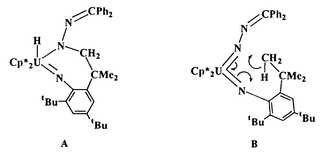
Clearly, the ability of the UN–R functionality to activate C–H bonds is related to the nature of the R group. For the imido ligand derived from diphenyldiazomethane, R is NCPh2, which, owing to the nitrogen, might be expected to provide more electron density to the imido nitrogen relative to previously reported uranium(VI) imido complexes in which R = alkyl or aryl.4–7,11,12 Accordingly, the imido nitrogen would experience enhanced basicity, which would translate into greater reactivity for the UN moiety.
In summary, this work demonstrates that through judicious choice of diazoalkane it is possible to enhance the reactivity of the UN linkage. Thus, diazoalkanes could serve as a versatile platform for the preparation of reactive uranium(VI) imido complexes. We are currently exploring the generality of this chemistry.
For financial support of this work, we acknowledge the Division of Chemical Sciences, Office of Basic Energy Sciences, U.S. Department of Energy (C. J. B.) and the LANL LDRD Program (D. E. M.). J. L. K. is the recipient of a Frederick Reines fellowship at Los Alamos. Finally, we thank Professor Paul B. Duval (U Missouri, Columbia) for helpful discussions.
Notes and references
- (a) P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729 CrossRef CAS; (b) C. C. Cummins, S. M. Baxter and P. T. Wolczanski, J. Am. Chem. Soc., 1988, 110, 8731 CrossRef CAS.
- (a) W. A. Nugent and B. L. Haymore, Coord. Chem. Rev., 1980, 31, 123 CrossRef CAS; (b) W. A. Nugent and J. M. Mayer, Metal–Ligand Multiple Bonds, Wiley, New York, 1988 Search PubMed; (c) D. E. Wigley, Prog. Inorg. Chem., 1994, 42, 239 Search PubMed; (d) P. Mountford, Chem. Commun., 1997, 2127 RSC; (e) J. L. Polse, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 13405 and references therein. Search PubMed.
- (a) G. K. Cantrell and T. Y. Meyer, J. Am. Chem. Soc., 1998, 120, 8035 CrossRef CAS; (b) R. L. Zuckerman, S. W. Krska and R. G. Bergman, J. Am. Chem. Soc., 2000, 122, 751 CrossRef CAS.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448 CrossRef CAS.
- R. C. Schnabel and C. J. Burns, unpublished results.
- R. G. Peters, B. P. Warner and C. J. Burns, J. Am. Chem. Soc., 1999, 121, 5585 CrossRef CAS.
- R. G. Peters, B. P. Warner, B. L. Scott and C. J. Burns, Organometallics, 1999, 18, 2587 CrossRef CAS.
- J. A. Davies, P. L. Watson, J. F. Liebman and A. Greenberg, Selective Hydrocarbon Activation: Principles and Progress, VCH, New York, 1990 Search PubMed.
- Experimental details are included as ESI†.
- M. Dartiguenave, M. J. Menu, E. Deydier, Y. Dartguenave and H. Siebald, Coord. Chem. Rev., 1998, 178–180, 623, and references therein Search PubMed.
- (a) C. J. Burns, W. H. Smith, J. C. Huffman and A. P. Sattelberger, J. Am. Chem. Soc., 1990, 112, 3237 CrossRef CAS; (b) D. S. J. Arney, C. J. Burns and D. C. Smith, J. Am. Chem. Soc., 1992, 114, 10068 CrossRef CAS; (c) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840 CrossRef CAS.
- B. P. Warner, B. L. Scott and C. J. Burns, Angew. Chem., Int. Ed., 1998, 37, 959 CrossRef CAS.
- S. Patai, The Chemistry of Diazonium and Diazo Groups, Parts 1 and 2, Wiley, New York, 1978, and references therein Search PubMed.
- SHELXTL/NT Version 5.1, Bruker AXS, Inc., Madison, WI.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental, including general procedures, materials and synthesis of complexes 2 and 3. See http://www.rsc.org/suppdata/cc/b1/b109455f/ |
‡ Crystal data for 2: C51H69N3U, M = 962.12, a = 12.407(4), b = 29.634(7), c = 12.836(4) Å, β = 103.484(5)°, V = 4590(2) Å3, monoclinic, space group P21/n, Z =4, T = 203 K, R1 (I > 2σ) = 0.0585, and wR2 (I > 2σ) = 0.1092.Crystal data for 3: C54H75N3U, M = 1004.20, a = 10.778(3), b = 21.982(7), c = 22.876(9) Å, α = 104.703(9), β = 98.253(6), γ
= 102.868(7)°, V = 4995(3) Å3, triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z =4, T = 203 K, R1 (I > 2σ) = 0.0951, and wR2 (I > 2σ) = 0.1153.The reflection data were collected on a Bruker P4/CCD using ϕ scans. The structure was solved using standard direct methods techniques (SHELXS-97),14 and refined using full-matrix least-squares based on F2 (SHELXL-97).14 Hydrogen atom positions were idealized, and rode on the atom they were attached to. All non-hydrogen atoms were refined anisotropically. CCDC reference numbers 172876 and 172877. See http://www.rsc.org/suppdata/cc/b1/b109455f/ for crystallographic data in CIF or other electronic format. , Z =4, T = 203 K, R1 (I > 2σ) = 0.0951, and wR2 (I > 2σ) = 0.1153.The reflection data were collected on a Bruker P4/CCD using ϕ scans. The structure was solved using standard direct methods techniques (SHELXS-97),14 and refined using full-matrix least-squares based on F2 (SHELXL-97).14 Hydrogen atom positions were idealized, and rode on the atom they were attached to. All non-hydrogen atoms were refined anisotropically. CCDC reference numbers 172876 and 172877. See http://www.rsc.org/suppdata/cc/b1/b109455f/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2002 |