
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn interrupted Corey–Chaykovsky reaction of designed azaarenium salts: synthesis of complex polycyclic spiro- and fused cyclopropanoids†
Bara
Singh‡
,
Arshad J.
Ansari‡
 ,
Nirmal
Malik
and
S. S. V.
Ramasastry
*
,
Nirmal
Malik
and
S. S. V.
Ramasastry
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector 81, Manauli PO, S. A. S. Nagar, Punjab 140306, India. E-mail: ramsastry@iisermohali.ac.in; ramsastrys@gmail.com; Web: https://web.iisermohali.ac.in/faculty/sastry/
First published on 1st June 2023
Abstract
Simultaneous dearomatizing spirannulation of pyridinium salts is still in its infancy. Here, we present an organized skeletal remodeling of designed pyridinium salts by utilizing an interrupted Corey–Chaykovsky reaction to access unprecedented and structurally intriguing molecular architectures such as the vicinal bis-spirocyclic indanones and spirannulated benzocycloheptanones. This hybrid strategy rationally merges the nucleophilic features of sulfur ylides with the electrophilic pyridinium salts to achieve the regio- and stereoselective synthesis of new classes of cyclopropanoids. The plausible mechanistic pathways were derived from experimental results and control experiments.
Introduction
Small organic molecules with broad scaffold diversity have found relevance in several drug discovery programs.1 The synthesis of privileged three-dimensional drug-like structures bears great potential for their ability to bind selectively to the desired target, and possess tunable physicochemical properties. Therefore, a synthetic challenge is to develop efficient methods to generate novel molecular architectures that can access untapped chemical space.Sulfur ylide chemistry has been demonstrated to be one of the mainstay tools adapted in the step- and pot-economic synthesis of complex structural motifs.2 In this context, it is worth mentioning the Corey–Chaykovsky (CC) reaction, which brings about the cyclopropanation of electron-deficient olefins by dimethyloxosulfonium methylide (DOSM), Scheme 1.3 The reaction involves displacing the dimethyl sulfoxide (DMSO) group from the zwitterionic intermediate A to generate cyclopropanes (path-a). In principle, A can be trapped by a suitable functionality (X = Y) to form various carbo- and heterocycles - the process we label as the interrupted CC reaction (path-b). This is the underlying theme of the present work.
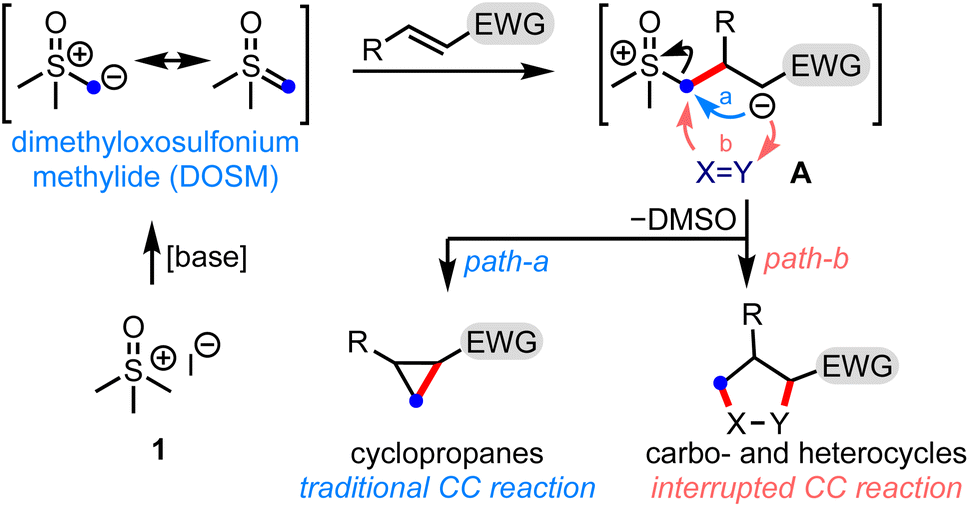 | ||
| Scheme 1 General representation of the Corey–Chaykovsky cyclopropanation and realization of a potential opportunity to interrupt the process. EWG = electron-withdrawing group. | ||
Just as sulfur ylides, pyridinium salts have manifested profound synthetic applications in natural products chemistry, pharmaceuticals, and material science.4 The inherent electrophilic nature of pyridinium salts facilitates nucleophilic additions at C-2/C-6 and C-4 positions, leading to the formation of dihydropyridines. During this process, the creation of an all-carbon quaternary center at C-4 can be envisioned, although sporadically pursued, Scheme 2a.5 In these lines, an ipso-nucleophilic addition of tethered nucleophiles at C-4 can be expected to create all-carbon spiro structures. But, the synthesis of spirocarbocyclic piperidines by this means is quite rarely accomplished.6,7 Interestingly, Eisch6a and Fraenkel6c achieved this synthetic feat through an intramolecular addition of hard nucleophiles such as organolithiums and Grignard reagents, although the C-4 position in pyridinium salts is soft. On the other hand, the synthetic utility of bis-enamine functionality of the so-formed dihydropyridines has also been underutilized.
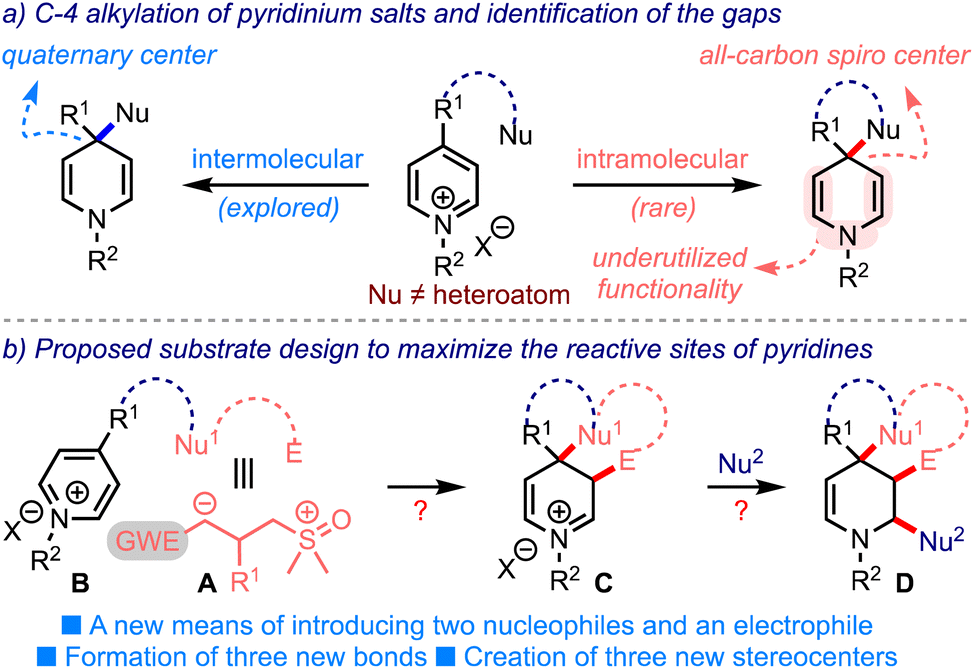 | ||
| Scheme 2 (a) The creation of an all-carbon quaternary/sipro center from pyridinium salts. (b) An outline of the proposed substrate design and an expected outcome. Nu = nucleophile. E = electrophile. | ||
With this background, we hypothesized an outline of the substrate design B as in Scheme 2b. We anticipated that rational incorporation of Nu1–E tether (with dual electronic characters such as A) onto the pyridinium ring would trigger the C-4 spirannulation. The resulting dienamine (not shown) could undergo an enamine-mediated alkylation, delivering the iminium species C. Subsequent intermolecular addition of an appropriate nucleophile (Nu2) could then furnish spiro-fused tetrahydropiperidines D incorporated with at least three new bonds and three new stereogenic centers. Thus, the proposed design efficiently utilizes a minimum of three of the five reactive sites of pyridinium salts.8
Results and discussion
In line with the hypothesis presented in Scheme 2b, a model substrate 2a was prepared by tethering the enone and pyridinium moieties ortho to each other on the arene backbone, Scheme 3. The reaction of 2a with DOSM in DMSO delivered the vicinal bis-spirocyclic indanones 3 as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 separable mixture of diastereomers, although in poor yield. The single-crystal X-ray diffraction analysis confirmed the structures, including the relative stereochemistry of 3a and 3a′.9 We were especially intrigued that a simple one-pot synthetic operation creates three new rings and three contiguous stereocenters, two of which are all-carbon vicinal spiro centers. As such, constructing a spiro structure is a synthetic challenge, even in the context of modern organic synthesis, and assembling vicinal spirocyclic systems is even more daunting.10 Considering the privileged status of indanes, cyclopropanes, and piperidines in drug discovery,11 the hybrid structure 3 and its analogs embodying these elements may serve as new pharmacophores in drug discovery. The encouraging features mentioned above inspired us to optimize the reaction conditions.
:1 separable mixture of diastereomers, although in poor yield. The single-crystal X-ray diffraction analysis confirmed the structures, including the relative stereochemistry of 3a and 3a′.9 We were especially intrigued that a simple one-pot synthetic operation creates three new rings and three contiguous stereocenters, two of which are all-carbon vicinal spiro centers. As such, constructing a spiro structure is a synthetic challenge, even in the context of modern organic synthesis, and assembling vicinal spirocyclic systems is even more daunting.10 Considering the privileged status of indanes, cyclopropanes, and piperidines in drug discovery,11 the hybrid structure 3 and its analogs embodying these elements may serve as new pharmacophores in drug discovery. The encouraging features mentioned above inspired us to optimize the reaction conditions.
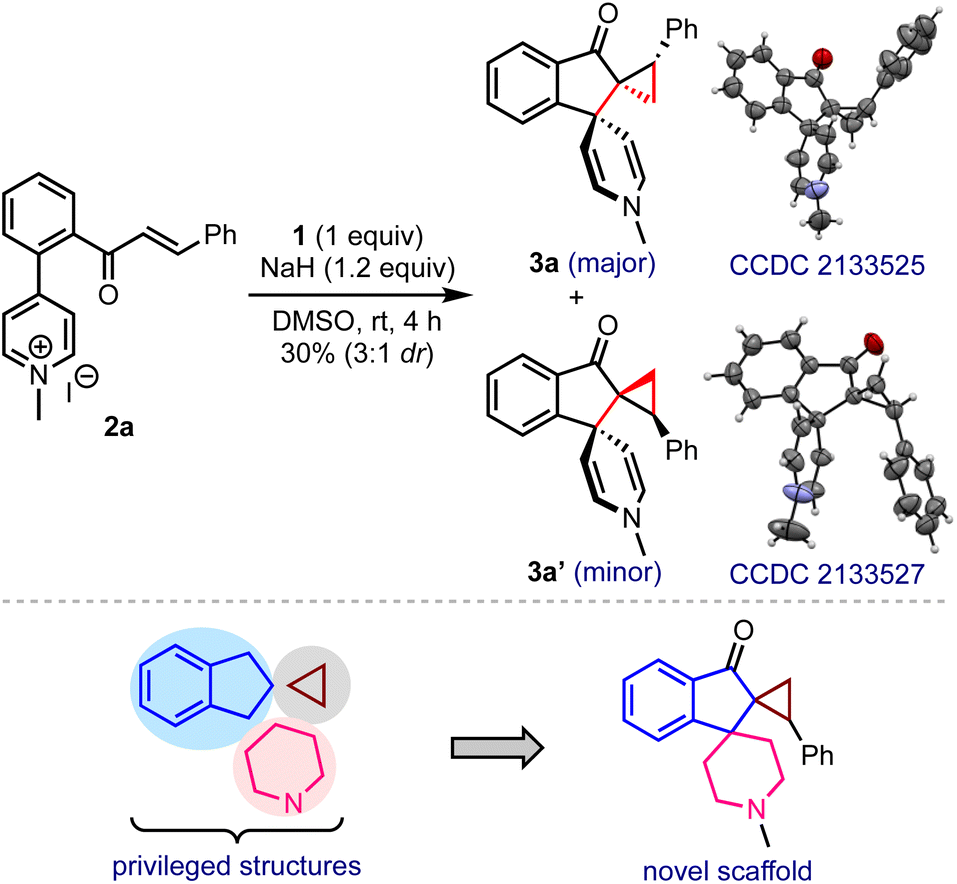 | ||
| Scheme 3 Hypothesis-driven substrate design and a preliminary result providing unprecedented vicinal bis-spirocyclic indanones. | ||
The initial solvent screening revealed a significant improvement in the efficiency of the reaction in DMF (entries 1–3, Table 1). The desired product 3 was obtained in 86% yield in a short reaction time. Subsequently, we evaluated the effect of the temperature. The reaction of 2a at 0 °C furnished 3 in moderate yield, whereas at 60 °C, the product was isolated in 75% yield (entries 4 and 5). Contrary to our expectation, we did not achieve a better result with the increased amounts of the base (entries 6 and 7). Our attempts to improve the yield by employing various inorganic or organic bases gave poor results (entries 8–12). Thus, a combination of NaH and DMF was suitable for an efficient outcome.
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yield of 3b [%] |
|
a See the ESI for a detailed procedure.
b Chromatographic yields.
c Obtained in 3.5:1 diastereomeric ratio (dr).
d At 0 °C.
e At 60 °C.
f In the presence of 1.5 equiv. of NaH.
g In the presence of 2 equiv. of NaH.
|
||||
| 1 | NaH | Acetamide | 24 | 23 |
| 2 | NaH | MeCN | 24 | 30 |
| 3c | NaH | DMF | 3 | 86 |
| 4d | NaH | DMF | 12 | 39 |
| 5e | NaH | DMF | 3 | 75 |
| 6f | NaH | DMF | 24 | 79 |
| 7g | NaH | DMF | 24 | 76 |
| 8 | KOtBu | DMF | 24 | 15 |
| 9 | CS2CO3 | DMF | 24 | — |
| 10 | K2CO3 | DMF | 24 | — |
| 11 | DBU | DMF | 24 | — |
| 12 | Guanidine | DMF | 24 | — |
The optimized conditions were applied to a wide range of enone-tethered pyridinium salts 2, and the results are compiled in Table 2. The electronic and steric roles of the substituents on the enone moiety (R1), arene backbone (R2), and pyridine core (R3 and R4) were thoroughly investigated.
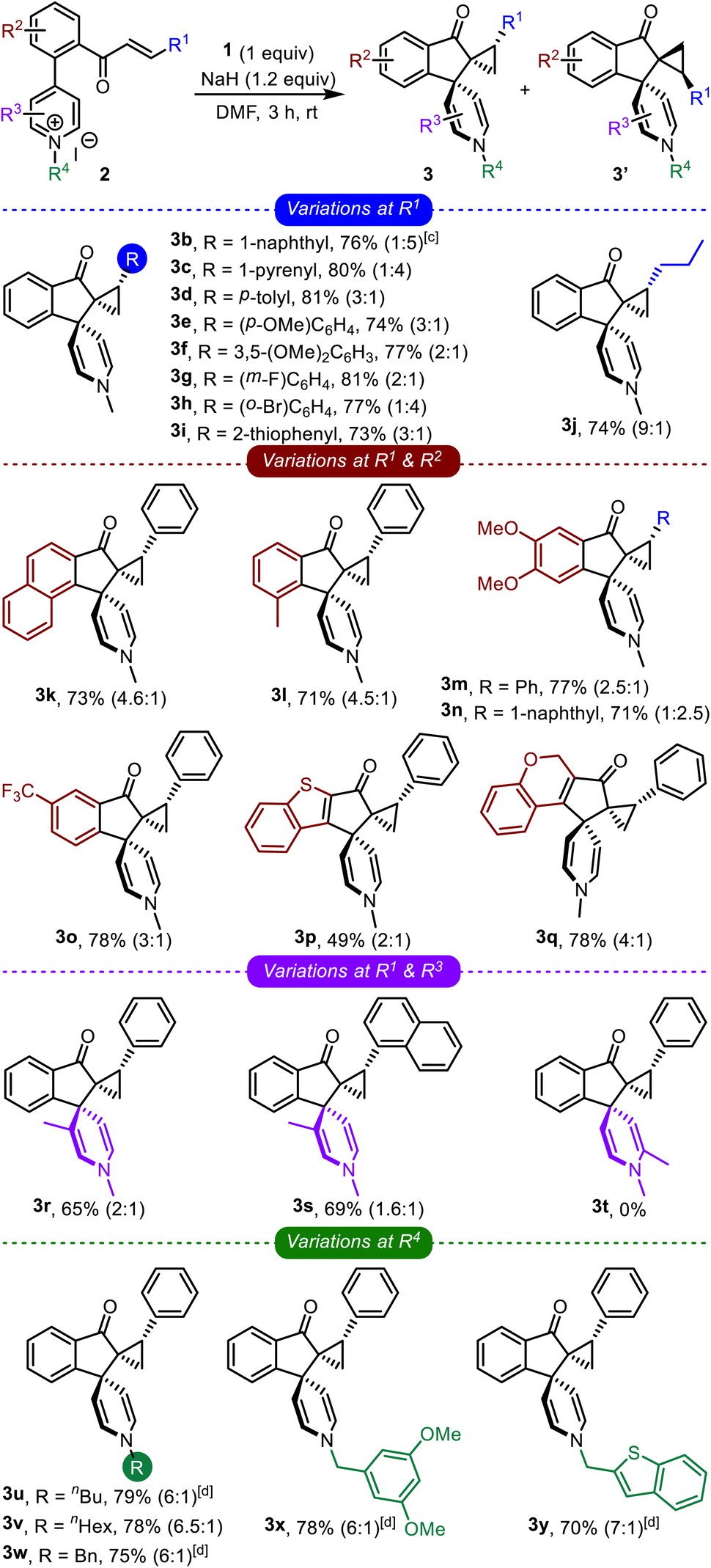
Different types of arenes possessing electron-donating as well as electron-withdrawing groups (3b–3h), heteroarenes (3i), and even alkyl groups (3j) were well-tolerated on the enone functionality and generated the respective bis-spirocyclic indanones in good yields, Table 2. Next, the role of the substituents on the arene backbone was studied. To that extent, the reaction was found to be quite efficient with substrates bearing the naphthalene backbone (3k), electron-rich arenes (3l–3n), CF3-substituted arenes (3o), heteroarenes such as benzothiophene (3p), and also with a non-aromatic backbone (3q). Although we were apprehensive about the fate of the C-3 substituted pyridinium salts, the respective products were still obtained in good yields (3r and 3s). However, surprisingly, the reaction of C-2 substituted pyridinium salts did not generate the desired product (3t).
Subsequently, we also investigated the role of N-substituents on the course of the reaction, Table 2. Various linear and branched alkyl systems were well-tolerated, giving the respective products in good yields (3u–3y). We observed that the diastereoselectivity improved (to 7:1) with the longer alkyl substituents. Next, the role of the counter anion (bromide instead of iodide) was also verified (3u and 3w–3y) and realized to be insignificant. Overall, the comprehensive study of the impact of R1, R2, R3, and R4 revealed that the transformation is quite robust, broad in scope, and capable of accommodating substituents with distinct electronic and steric characters.
To extend the scope of the method, we attempted the reaction of the biaryl pyridinium salt possessing the β,β-disubstituted enone system (2z), Scheme 4. To our delight, the expected indanone 3z was isolated in good yield despite the steric encumbrance at the β-position. This result demonstrates the feasibility of creating three contiguous quaternary carbons, two of which are spiro centers.
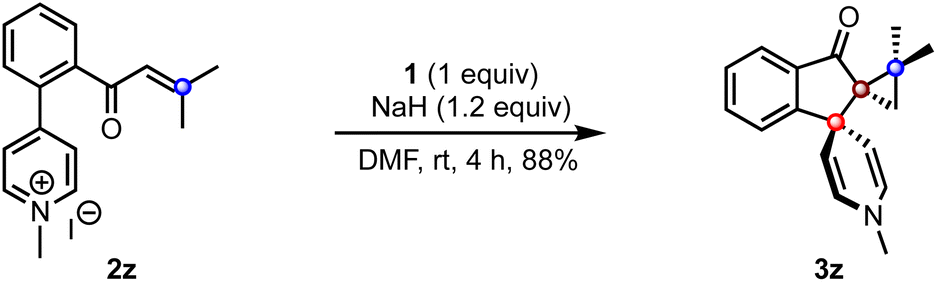 | ||
| Scheme 4 The reaction of β,β-disubstituted enones to create three contiguous quaternary centers. | ||
While evaluating the substrate scope, we questioned whether the enone-tethered quinolinium salts such as 4a could be tolerated under the reaction conditions and generate the expected product 3aa, Scheme 5. Interestingly, the reaction of 4a and 1 under the optimized conditions delivered an unexpected product 5a in 78% yield as a single isomer. The structure and the relative stereochemistry of 5a were confirmed from the single-crystal X-ray diffraction analysis.12 We were surprised by the formation of three new rings with an unusual 6–3–7 fusion, four new C–C bonds, five contiguous stereocenters, and a fully-substituted quinoline core incorporated with a methylene sulfoxide moiety in a simple one-step operation. The unprecedented pentacyclic scaffold 5 can be envisioned as a combination of (benzo)cycloheptanes, (tetrahydro)quinolines, and cyclopropanes, which are commonly encountered in several natural products and drug candidates.13 With the belief that this hybrid motif finds applications in devising novel classes of bioactive molecules and becomes a new template in drug discovery, we examined the generality of the method.
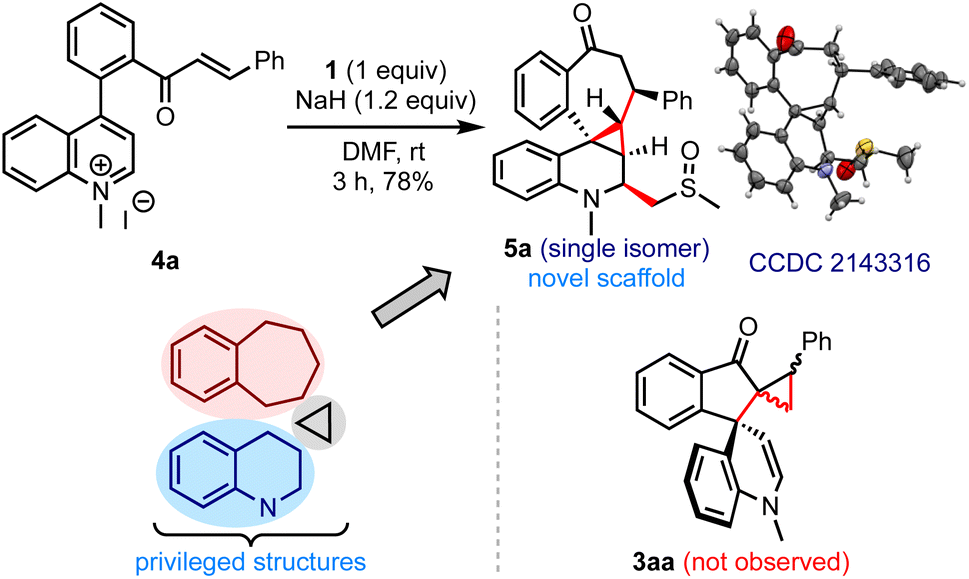 | ||
| Scheme 5 An unexpected result obtained with enone-tethered quinolinium salts: access to a new class of polycyclic benzocycloheptanones. | ||
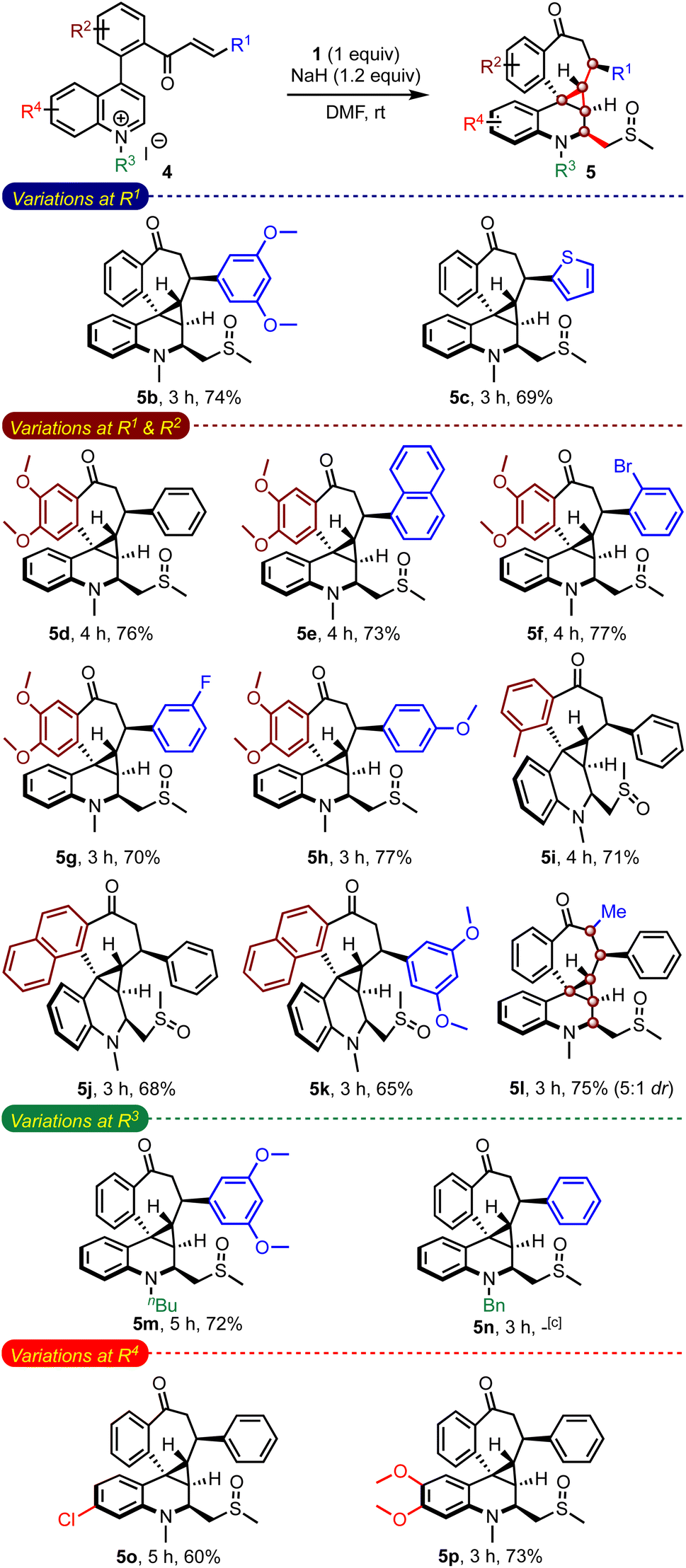
An evaluation of the substrate scope with an array of enone-tethered quinolinium salts 4 revealed the generality of the method in assembling a range of complex spiro-fused benzocycloheptanones 5b–5n, Table 3.14 The role of the substituents on the enone moiety (R1) and the arene backbone (R2), and N-substituents (R3) was studied in detail. For example, an arene ring with electron-donating methoxy groups at R1 gave the expected product 5b in good yield, although unfavourable for the initial Michael addition of the sulfur ylide. Even heteroarene moieties were also tolerated at R1 (5c). We also evaluated the substrates with methoxy and methyl groups on the arene backbones (R2) while varying substituents at R1 (5d–5i). All of them fared consistently well under the reaction conditions. Natural product-like hexacyclic benzocycloheptanone derivatives such as 5j and 5k could also be efficiently synthesized. Of particular interest, the reaction of α,β-substituted enone-quinolinium salt 4l resulted in forming 5l with six contiguous stereocenters. On the other hand, the reaction of the n-butyl quinolinium salt delivered the respective product 5m in good yield, but the N-benzyl salt did not generate the expected product 5n. We also evaluated the role of substituents on the quinoline portion (R4). Accordingly, we prepared complex pentacyclic structures with chloro (5o) and dimethoxy (5p) groups. A narrow yield range (60–77%) across a wide range of substrates is indicative of the robustness of the method.
Next, we intended to develop a one-pot synthesis of bis-spirocyclic indanones 3 starting from biaryl pyridine-enones 6, avoiding the pre-formation of the salts 2, Scheme 6. Accordingly, 6a and 6b were treated with methyl iodide in DMF. After ensuring their conversion to the respective pyridinium salts, 2a and 2b, they were treated with DOSM and obtained 3a and 3b, in 28% and 25% yields, respectively. However, when the salt-making step was performed in DCM, the overall yields improved significantly (to 59% and 57%, respectively). This was attributed to the high solubility of the pyridinium salts 2 in DCM. Subsequently, we extended this strategy to synthesizing spiro-fused benzocycloheptanones 5 in one-pot, starting from the respective biaryl quinolines 7. Thus, 5f and 5m were obtained in 61% and 64% yields from 7f and 7m, respectively, even when performed on 1 mmol scales.
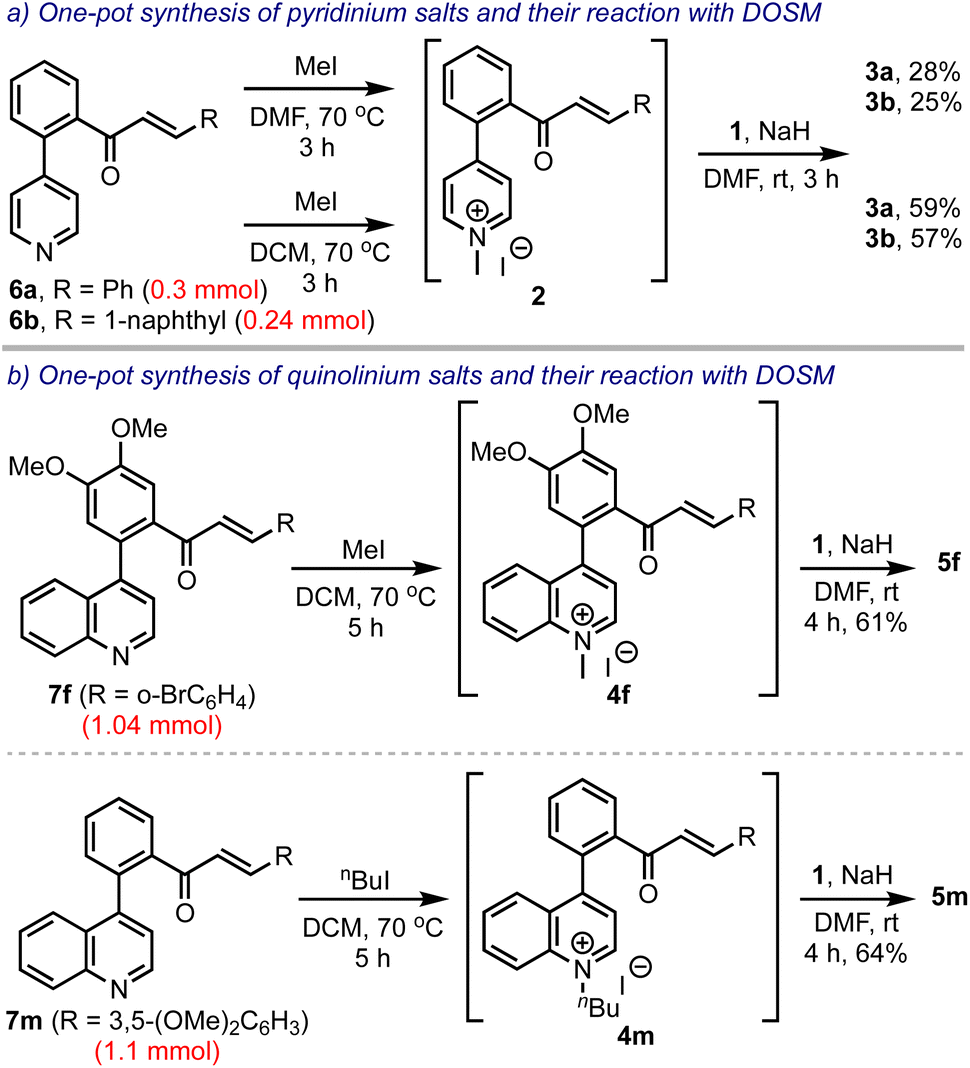 | ||
| Scheme 6 One-pot synthesis of bis-spirocyclic indanones and spiro-fused benzocycloheptanones. | ||
Prior to arriving at the plausible mechanistic pathways leading to the product formation, we conducted a few control experiments, Scheme 7. The reaction of 2a was performed with an in situ generated trideuteromethyl sulfoxonium iodide (TDMSOI)15 and obtained the respective product 3a-D with ∼40% deuterium incorporation at the methylene group of the cyclopropane moiety, Scheme 7a. The simple biaryl pyridine-enone 6a, when treated with DOSM, gave only the cyclopropyl-ketone 8a, Scheme 7b. Interestingly, the pyridinium salt 9a lacking the enone portion remained as such under the reaction conditions, Scheme 7c. The reaction of 2a was conducted in the presence of DOSM, and the proton sponge still generated 3a in 78% yield, indicating that the reaction generated no acid and it was not responsible for facilitating any transformation, Scheme 7d. Finally, we also evaluated the reaction of 2a with trimethylsulfonium iodide and sodium hydride, but surprisingly, it did not yield any product, Scheme 7e.
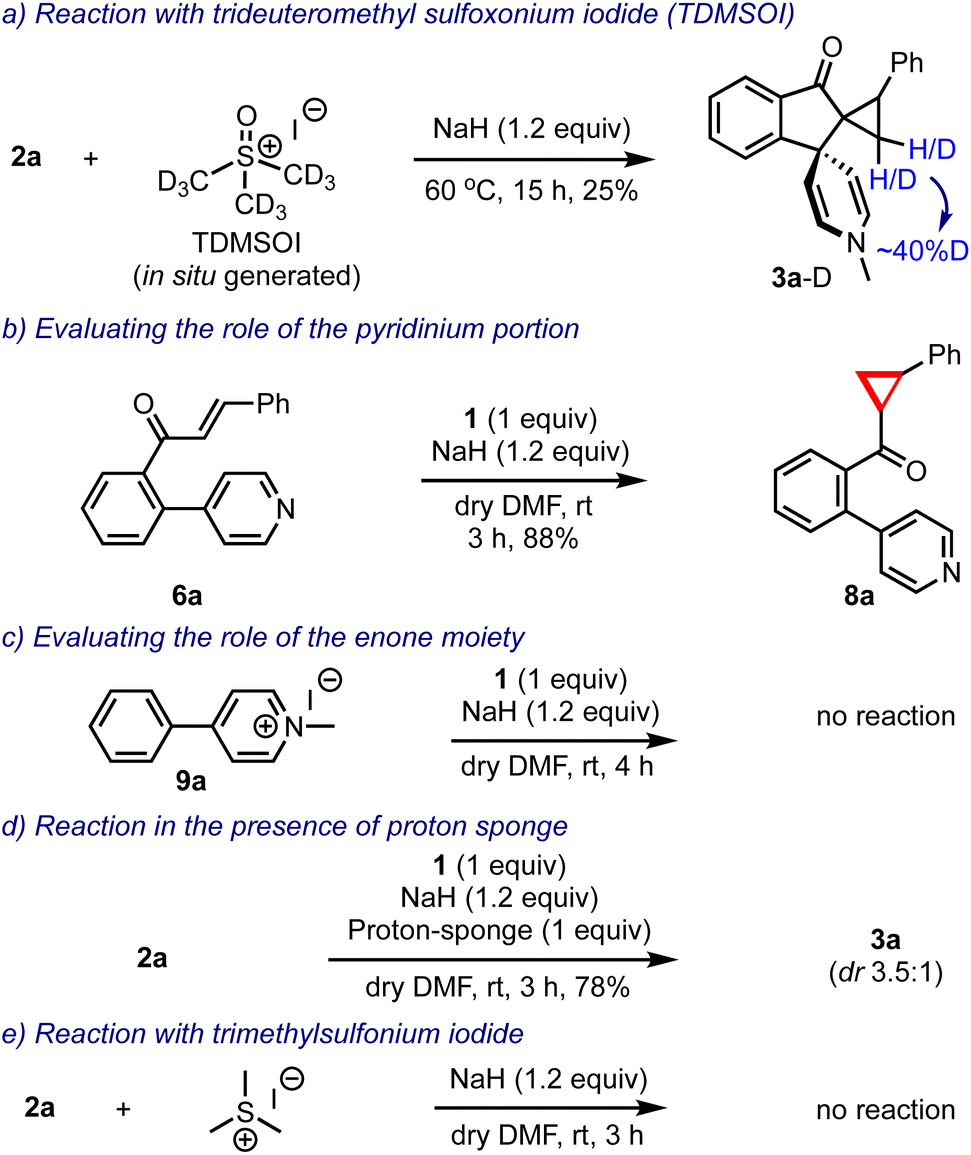 | ||
| Scheme 7 Control experiments performed to gain mechanistic insights. | ||
Based on the experimental results and control experiments, a plausible mechanism is proposed in Scheme 8. The reaction commences with the Michael addition of DOSM to 2 or 4, leading to the formation of enolates E, which, in the case of pyridinium salts, react in an aldol fashion from the C-4 position to generate spiroindanones F.16 Eventually, the bis-spirocyclic indanones 3 are produced via enol-assisted displacement of the DMSO. On the other hand, in the case of quinolinium salts, a 1,3-/1,5-proton shift of E provides ylides H, which undergo C-4 addition to generate the key intermediate I. An enamine-assisted displacement of the DMSO follows the entrapment of iminium ions J with sulfoxide to form K. Subsequent [2,3]-O-to-C rearrangement of ylides L provides 5.17
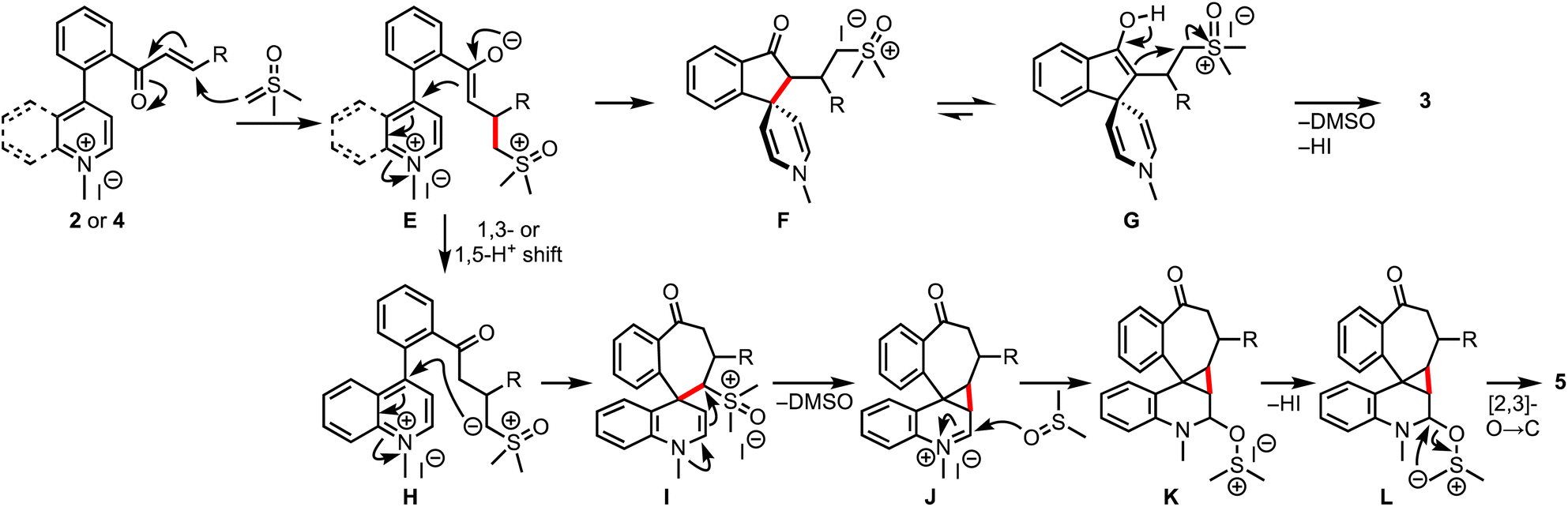 | ||
| Scheme 8 Plausible mechanism for the formation of 3 and 5. | ||
Conclusions
We demonstrated an interrupted Corey–Chaykovsky reaction of designed enone-tethered azaarenium salts to access complex spiro- and fused cyclopropanoids, which are difficult to access via conventional synthetic transformations. This method also provided rapid and efficient access to structurally and electronically diverse unprecedented scaffolds such as vicinal bis-spirocyclic indanones 3 and spirannulated polycyclic benzocycloheptanones 5. We also addressed several important questions surrounding the mechanism of the divergent pathways from detailed experimental studies. Some other salient features of this study are: (i) it represents the first stereoselective transformation of sulfoxonium ylides and pyridinium salts, and (ii) it establishes a new substrate-based folding pathway. The synthetic methods described herein hold great potential and will stimulate further research in synthesizing novel carbo- and heterocycles from easily accessible pyridine derivatives. Efforts to extend the concepts to new substrate classes are in progress, and the details will be communicated in due course.Data availability
Data supporting this article have been uploaded as ESI.†Author contributions
B. S. and A. J. A. conducted the investigation under S. S. V. R.'s supervision. B. S., A. J. A. and S. S. V. R. wrote the manuscript. B. S., A. J. A. and N. M. prepared the ESI.† All authors contributed to the editing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank IISER Mohali for the NMR, mass spectrometry, and X-ray facilities. S. S. V. R. thanks SERB for the Swarnajayanti fellowship (DST/SJF/CSA-01/2017–18) and IISER Mohali for funding. B. S. thanks UGC for a research fellowship and N. M. thanks the Ministry of Education for the Prime Minister's Research Fellowship (PMRF). A. J. A. thanks IISER Mohali for a postdoctoral fellowship.Notes and references
- (a) B. R. Stockwell, Nature, 2004, 432, 846–854 CrossRef CAS PubMed; (b) M.-Q. Zhang and B. Wilkinson, Curr. Opin. Biotechnol., 2007, 18, 478–488 CrossRef CAS PubMed; (c) S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6699–6702 CrossRef CAS PubMed; (d) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed; (e) S. E. Dalton and S. Campos, ChemBioChem, 2020, 21, 1080–1100 CrossRef CAS PubMed.
- (a) A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341–2372 CrossRef CAS PubMed; (b) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (c) P. Li, Synlett, 2021, 32, 1275–1280 CrossRef CAS.
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353 CrossRef CAS; (b) Y. G. Gololobov, A. N. Nesmeyanov, V. P. Lysenko and I. E. Boldeskul, Tetrahedron, 1987, 43, 2609–2615 CrossRef CAS; (c) M. M. Heravi, S. Asadi, N. Nazari and B. M. Lashkariani, Curr. Org. Synth., 2016, 13, 308–333 CrossRef CAS.
- Selected reviews: (a) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453–493 RSC; (b) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943–8960 CrossRef CAS; (c) S. L. Rossler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreirs and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264–9280 CrossRef PubMed.
- (a) M. W. Gribble, S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057–5060 CrossRef CAS PubMed; (b) Y. Yang, C.-H. Xu, Z.-Q. Xiong and J.-H. Li, Chem. Commun., 2020, 56, 9549–9552 RSC; (c) C.-H. Xu, J.-H. Li, J.-N. Xiang and W. Deng, Org. Lett., 2021, 23, 3696–3700 CrossRef CAS PubMed.
- For pyridine C4-carbospirannulations, see: (a) J. J. Eisch, C. A. Kovacs, P. Chobe and M. P. Boleslawski, J. Org. Chem., 1987, 52, 4427–4437 CrossRef CAS; (b) T. Muramatsu, A. Toyota and M. Suzuki, J. Am. Chem. Soc., 2005, 127, 4572–4573 CrossRef CAS PubMed; (c) G. Fraenkel, A. Chow, Y. Liang, J. Song and J. A. Gallucci, Helv. Chim. Acta, 2012, 95, 2063–2071 CrossRef CAS; (d) R.-J. Yan, B.-X. Xiao, Q. Ouyang, H.-P. Liang, W. Du and Y.-C. Chen, Org. Lett., 2018, 20, 8000–8003 CrossRef CAS PubMed; (e) J. C. Abell, C. P. Bold, L. Vicens, T. Jentsch, N. Velasco, J. L. Tyler, R. N. Straker, A. Noble and V. K. Aggarwal, Org. Lett., 2023, 25, 400–404 CrossRef CAS PubMed.
- For pyridine C4-heterospirannulations, see: (a) D. D. Weller, G. R. Luellen and D. L. Weller, J. Org. Chem., 1983, 48, 3061–3067 CrossRef CAS; (b) J. Clayden, S. D. Hamilton and R. T. Mohammed, Org. Lett., 2005, 7, 3673–3676 CrossRef CAS PubMed; (c) G. Arnott, H. Brice, J. Clayden and E. Blaney, Org. Lett., 2008, 10, 3089–3092 CrossRef CAS PubMed; (d) S. G. Parameswarappa and F. C. Pigge, Org. Lett., 2010, 12, 3434–3437 CrossRef CAS PubMed.
- Some interesting strategies to functionalize pyridinium salts, see: (a) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed; (b) O. G. Mancheno, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823–8827 CrossRef PubMed; (c) G. Bertuzzi, A. Sinisi, L. Caruana, A. Mazzanti, M. Fochi and L. Bernardi, ACS Catal., 2016, 6, 6473–6477 CrossRef CAS; (d) G. Bertuzzi, L. Bernardi and M. Fochi, Catalysts, 2018, 8, 632 CrossRef; (e) J. Lee, D. Ko, H. Park and E. J. Yoo, Chem. Sci., 2020, 11, 1672–1676 RSC; (f) H.-J. Miao, L.-L. Wang, H.-B. Han, Y.-D. Zhao, Q.-L. Wang and Z.-W. Bu, Chem. Sci., 2020, 11, 1418–1424 RSC; (g) X.-G. Bai, H.-J. Miao, Y. Zhao, Q.-L. Wang and Z.-W. Bu, Org. Lett., 2020, 22, 5068–5073 CrossRef CAS PubMed; (h) X. Song, R.-J. Yan, W. Du and Y.-C. Chen, Org. Lett., 2020, 22, 7617–7621 CrossRef CAS PubMed; (i) D. J. Robinson, S. P. Spurlin, J. D. Gorden and R. R. Karimov, ACS Catal., 2020, 10, 51–55 CrossRef CAS; (j) T.-T. Li, Y. You, T.-J. Sun, Y.-P. Zhang, J.-Q. Zhao, Z.-H. Wang and W.-C. Yuan, Org. Lett., 2022, 24, 5120–5125 CrossRef CAS PubMed; (k) L.-J. Gu, H.-B. Han, Z.-W. Bu and Q.-L. Wang, Org. Lett., 2022, 24, 2008–2013 CrossRef CAS PubMed.
- X-ray crystallographic data of compounds 3a and 3a′ are provided in the ESI. CCDC 2133525 (3a) and 2133527 (3a′).†.
- (a) W.-L. Chan, X. Tang, F. Zhang, G. Quek, G.-J. Mei and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 6260–6264 CrossRef CAS PubMed; (b) J. Zhang, W.-L. Chan, L. Chen, N. Ullah and Y. Lu, Org. Chem. Front., 2019, 6, 2210–2214 RSC; (c) C. Wang, D. Wen, H. Chen, Y. Deng, X. Liu, X. Liu, L. Wang, F. Gao, Y. Guo, M. Sun, K. Wang and W. Yan, Org. Biomol. Chem., 2019, 17, 5514–5519 RSC; (d) W. Luo, B. Shao, J. Li, X. Xiao, D. Song, F. Ling and W. Zhong, Org. Chem. Front., 2020, 7, 1016–1021 RSC.
- (a) M. Vilums, J. Heuberger, L. H. Heitman and A. P. Ijzerman, Med. Res. Rev., 2015, 35, 1097–1126 CrossRef CAS PubMed; (b) T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed; (c) A. Peneau, P. Retailleau, C. Guillou and L. Chabaud, J. Org. Chem., 2018, 83, 2324–2340 CrossRef CAS PubMed; (d) M. M. Abdelshaheed, I. M. Fawzy, H. I. El-Subbagh and K. M. Youssef, Future J. Pharm. Sci, 2021, 7, 188–198 CrossRef.
- X-ray crystallographic data of compounds 5a is provided in the ESI. CCDC 2143316 (5a).†.
- (a) K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS PubMed; (b) R. Musiol, K. Malarz and J. Mularski, Curr. Org. Chem., 2017, 21, 1896–1906 CrossRef CAS; (c) J.-H. Fan, Y.-J. Hu, L.-X. Li, J.-J. Wang, S.-P. Li, J. Zhao and C.-C. Li, Nat. Prod. Rep., 2021, 38, 1821–1851 RSC; (d) A. Mondal, Shivangi, P. Tung, S. V. Wagulde and S. S. V. Ramasastry, Chem. Commun., 2021, 57, 9260–9263 RSC.
- The lifetime of the products was found to be short (of the range of 5-8 days) even when refrigerated.
- (a) Z. Shen, S. Zhang, H. Geng, J. Wang, X. Zhang, A. Zhou, C. Yao, X. Chen and W. Wang, Org. Lett., 2019, 21, 448–452 CrossRef CAS PubMed; (b) M.-H. Zhu, C.-L. Yu, Y.-L. Feng, M. Usman, D. Zhong, X. Wang, N. Nesnas and W.-B. Liu, Org. Lett., 2019, 21, 7073–7077 CrossRef CAS PubMed.
- Owing to design considerations, the reaction at C-2/C-6 position is highly unlikely.
- A. Kobayashi, T. Matsuzawa, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 5429–5432 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2133525 (3a), 2133527 (3a′) and 2143316 (5a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01578e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |