Determination of trace concentrations of hexavalent chromium
Michael
Gardner
* and
Sean
Comber
WRc-NSF, Henley Road, Medmenham, Marlow, Buckinghamshire, UK SL7 2HD. E-mail: gardner_mj@wrcplc.co,uk; Fax: + 44 1491 579094; Tel: + 44 1491 636555
First published on 11th December 2001
Abstract
A simple and sensitive solvent extraction-atomic spectrometric technique has been developed for the determination of hexavalent chromium in fresh and saline waters. The technique is based on the reaction of chromium with diphenylcarbazide. The method has been tested on a variety of water samples over an analytical range of 0–2 μg l−1. A limit of detection of 0.024 μg l−1 was achieved. Spiking recoveries in the range 87–115% were achieved in river water, drinking water and marine waters.
Introduction
Metal speciation is usually the key to the fate and behaviour of metals.1 In the case of chromium, the important issue is the relative proportions of the trivalent (CrIII) and hexavalent CrVI forms. The biological effects of the metal in the two oxidation states are markedly different. The trivalent form is relatively non-toxic and is regarded as an essential trace element, whilst CrVI is of relatively high toxicity and has been shown to be a carcinogen in animal studies.Public concern has been expressed in the US in relation to possible exposure of drinking water consumers to hexavalent chromium. In 1999, the US Office of Environmental Health Hazard Assessment of the Environment Protection Agency established a public health goal of 2.5 μg l−1 for total chromium, based on a health protective level of 0.2 μg l−1 for hexavalent chromium (derived for a cancer endpoint) and the assumption that the hexavalent chromium is no more than 7% of the total chromium. However, a limited study of drinking water sources conducted in late 1999 indicated that the average percentage of hexavalent chromium may be above 50%.2 At present, in the UK and other EU states concentrations of total chromium in drinking water are monitored for compliance with a limit concentration of 50 μg l−1. The UK environmental quality standard for total chromium in surface waters is set at 15 μg l−1, though there may be concern about lower concentrations if the metal were present as the hexavalent form. A reduction in the concentration of interest and a focus on CrVI species generates a requirement for analytical methodology suitable for monitoring purposes in both drinking waters and surface waters. This paper describes a procedure, which was developed and tested with the aim of meeting this requirement.
Methodology
Analytical techniques available for the determination of chromium speciation at trace levels include electrochemical methods (e.g. stripping voltammetry) and methods involving separation of species and subsequent determination of the separated fractions using an analytical technique for total metal. Separation may be on to a solid phase,3,4 or into a solvent.5,6 There is usually a need to achieve some degree of preconcentration, so that the analytical method is capable of determining suitably low concentrations of chromium. Electrochemical methods are capable of measuring naturally occurring chromium concentrations and species directly.7 However, the methodology tends to be complex and lacking in robustness. Several separation methods with adequate detection capability for the determination of total chromium and CrIII have been reported8,9 but none provides a simple means of determining CrVI directly. Chromium(VI) concentrations may be arrived at by subtraction of the concentration of CrIII from total dissolved Cr. This subtraction may be subject to large uncertainty, particularly if CrIII is the predominant form. Furthermore these methods tend to be complicated and expensive to apply. The method recommended by legislators in the US for the determination of CrVI is one in which CrVI is isolated by liquid chromatography and determined colorimetrically. This technique has a reported limit of detection of 0.5 μg l−1.10 The use of a technique with a limit of detection so close to the proposed water quality standard value may impose a serious limitation on the ability to monitor and regulate levels of chromiumVI.The methodology for the determination of CrVI by colorimetry using diphenylcarbazide is well established.11 Direct spectrophotometry can be used to determine CrVI in clean waters down to a limit of detection of approximately 2–3 μg l−1. This is not adequate to monitor compliance with quality standards or limit values set at the low levels discussed above. This work aimed to develop the diphenylcarbazide methodology for the determination of CrVI, principally by extending it to sub-microgram per litre levels using preconcentration by solvent extraction.
Diphenylcarbazide gives a sensitive and specific colour reaction with CrVI in mineral acid solution. The pink coloured chromophore is a chelate of CrIII and diphenylcarbazone. The latter is produced and simultaneously combines with chromium when diphenylcarbazide is oxidised by CrVI. The reaction may be summarised as:
| 2CrO42− + 3H4L + 8H+ = [CrIII (HL)2]+ + Cr3+ + H2L + 8H2O |
and H2L diphenylcarbazone:
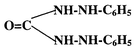
Direct reaction of CrIII with diphenylcarbazone does not occur to any appreciable extent on account of the well known inertness of the CrIII aquo-complex. The singly charged chromium–diphenylcarbazone complex can be extracted into relatively polar solvents as an ion pair with chloride ion. Sandell and Onishi12 suggest isoamyl alcohol as a suitable solvent.
Target analytical performance
The following performance criteria were defined13,14,15 as desirable in a method to be used for monitoring of CrVI in drinking waters and surface waters, assuming that it is necessary to operate in the range 0–2 μg l−1 and that the establishment of compliance with a quality standard of 2 μg l−1 is of primary interest. The total standard deviation of individual results should be less than 5% of the determinand concentration or 0.01 μg l−1, whichever was the larger; spiking recovery (both saline and fresh water samples) should not be significantly outside the range 90–110%; the limit of detection should be 0.03 μg l−1 or better.Experimental
Reagents
Diphenylcarbazide reagent, 0.25 g of diphenylcarbazide (Sigma Chemicals) was dissolved in 25 ml acetone (prepared freshly each day); sulfuric acid, 5 M: concentrated sulfuric acid (98% w/w) was diluted 3.5 fold with deionised water; iso amyl alcohol (Merck); saturated solution of sodium chloride, 300 g l−1. CrVI standards were prepared by diluting Merck Spectrosol (1000 mg l−1) standards with deionised water. CrIII standards were prepared from potassium chromium sulfate (Merck).All water was deionised and all chemicals were of reagent grade. All apparatus was pre-soaked in 5% v/v nitric acid and rinsed with deionised water before use.
Procedure
25 ml of sample was transferred to a graduated 50 ml polypropylene screw capped tube. To this was added 0.25 ml of 5 M sulfuric acid and 0.5 ml of diphenyl carbazide reagent. The sample was swirled to mix and left for10 min to allow colour development. Then 20 ml of saturated sodium chloride solution was added, followed by 2.5 ml of isoamyl alcohol. The tube was capped and shaken for 4 min.Extractions were performed in batches of 24 samples with the sample tubes held and shaken in a laboratory tube rack. After leaving the samples for at least half an hour for the solvent layer to separate, 0.5 ml of the upper alcohol layer was pipetted off and transferred for analysis by electrothermal atomic absorption spectrophotometry. Standard solutions in deionised water at concentrations of 0, 0.1, 0.5, 1 and 2 μg l−1 were extracted along with samples. All determinations were made on a Perkin-Elmer 4000 atomic absorption spectrometer and HGA 400 atomiser at 357.9 nm, with a 0.7 nm bandpass. Background correction was carried out using a deuterium lamp. The furnace programme used is shown in Table 1.
For the purpose of the performance tests reported here, five different water samples were analysed in duplicate, unspiked and spiked with CrVI at 0.5 and 2 μg l−1 over a set of nine analytical runs (see Table 2. All test samples were prepared in bulk by filtration under positive nitrogen pressure through acid-washed cellulose acetate filters (0.45 μm, 47 mm (Sartorius, Watford, UK)).
| Units | River water A | River water B | Estuarine sample C | Seawater sample D | Drinking water E | |
|---|---|---|---|---|---|---|
| a nd = not determined. na = not applicable. | ||||||
| Calcium | mg l−1 | 2.5 | 96 | 189 | 360 | 110 |
| Sodium | mg l−1 | nd | nd | 3489 | 10157 | 20 |
| Chloride | mg l−1 | nd | nd | 6268 | 18247 | 13 |
| Magnesium | mg l−1 | 2 | 4.4 | 415 | 1200 | 20 |
| Potassium | mg l−1 | nd | nd | 122 | 351 | 3 |
| DOC | mg l−1 | 12.1 | 5.1 | 4.2 | <1 | <1 |
| pH | 4.3 | 8.1 | 8 | 8 | 7.7 | |
| Electrical conductivity | μS cm −1 | 142 | 573 | na | na | 650 |
| Salinity | ppt | na | na | 12 | 35 | na |
Results
Fig. 1 shows a typical calibration curve, to which a quadratic fit is appropriate to at least 2 μg l−1, with a consistent reagent blank corresponding to approximately 0.02 μg l−1. The response of the technique to CrIII was tested for a blank sample spiked at CrIII concentrations of 0, 1, 5, 10, 20 and 50 μg l−1. The results, expressed as observed CrVI, were not statistically significantly different (p = 0.05) from zero and ranged between −0.010 and 0.017 μg l−1. The principal performance test characteristics required by UK regulatory agencies for water analysis13,14 for the six test samples are shown in Table 3. These include within run, between run and total standard deviation, spiking recovery and limit of detection. Performance test results (Table 3), based on 9 batches of analysis, showed that the precision and recovery achieved by the method met the chosen performance criteria.![Typical calibration graph showing response in milli absorbance units (mAU) versus concentration of CrVI in μg l−1. The equation of the quadratic curve fitted to the points is y = 36.6x2 + 437x−1.6, where y is mAU and x is [CrVI] in μg l−1. Error bars are 95% confidence limits based on 4 replicate measurements at each concentration.](/image/article/2002/AN/b109374f/b109374f-f1.gif) | ||
| Fig. 1 Typical calibration graph showing response in milli absorbance units (mAU) versus concentration of CrVI in μg l−1. The equation of the quadratic curve fitted to the points is y = 36.6x2 + 437x−1.6, where y is mAU and x is [CrVI] in μg l−1. Error bars are 95% confidence limits based on 4 replicate measurements at each concentration. | ||
| Sample (+ spike concentration μg l−1) | Mean value | Within run sa | Between run sb | Total sc | RSD (%) | DFd | Recovery (%) | Recovery confidence limits +/−e |
|---|---|---|---|---|---|---|---|---|
| a Within run s = within run standard deviation estimated as a pooled value with 9 degrees of freedom. b Between run s = between run standard deviation estimated with 8 degrees of freedom. c Total s = total standard deviation estimated as the combination of (a) and (b) with degrees of freedom indicated (see ref. 14, Cheeseman et al., 1989). d Degrees of freedom associated with the estimated total standard deviation (see note c above). e 95% confidence limits associated with the observed recovery. | ||||||||
| Sample A | 0.064 | 0.020 | 0.024 | 0.031 | 49 | 12 | ||
| Sample A + 0.5 μg l−1 | 0.50 | 0.025 | 0.018 | 0.031 | 6 | 15 | 87 | 4 |
| Sample A + 2 μg l−1 | 2.03 | 0.061 | 0.000 | 0.061 | 3 | 17 | 98 | 1 |
| Sample B | 0.15 | 0.013 | 0.012 | 0.017 | 11 | 13 | ||
| Sample B + 0.5μg l−1 | 0.66 | 0.018 | 0.030 | 0.035 | 5 | 10 | 101 | 5 |
| Sample B + 2 μg l−1 | 2.10 | 0.047 | 0.065 | 0.080 | 4 | 11 | 97 | 3 |
| Sample C | 0.17 | 0.013 | 0.017 | 0.021 | 12 | 11 | ||
| Sample C + 0.5 μg l−1 | 0.75 | 0.042 | 0.048 | 0.064 | 8 | 12 | 115 | 8 |
| Sample C + 2 μg l−1 | 2.17 | 0.038 | 0.081 | 0.089 | 4 | 10 | 100 | 3 |
| Sample D | 0.16 | 0.012 | 0.025 | 0.027 | 17 | 10 | ||
| Sample D + 0.5 μg l−1 | 0.72 | 0.029 | 0.053 | 0.060 | 8 | 10 | 112 | 8 |
| Sample E + 2 μg l−1 | 2.23 | 0.063 | 0.135 | 0.149 | 7 | 10 | 104 | 5 |
| Sample E | 0.086 | 0.005 | 0.008 | 0.010 | 12 | 11 | ||
| Sample E + 0.5 μg l−1 | 0.59 | 0.023 | 0.019 | 0.030 | 5 | 14 | 101 | 4 |
| Sample E + 2 μg l−1 | 2.06 | 0.042 | 0.091 | 0.100 | 5 | 10 | 99 | 3 |
| Standard solutions | ||||||||
| 0.1 μg l−1 | 0.10 | 0.016 | 12 | |||||
| 0.5 μg l−1 | 0.51 | 0.019 | 12 | |||||
| 1 μg l−1 | 1.00 | 0.022 | 12 | |||||
| 2 μg l−1 | 2.01 | 0.010 | 12 | |||||
| Limit of detection = 0.024 μg l−1 | ||||||||
Spiking the water samples at 0.5 and 2 μg l−1 made it possible to assess potential matrix interferences by comparison of the calibration slope for the natural water samples with that obtained for the standard solutions. Fig. 2 shows the ratio of calibration slope in the sample matrix versus that in deionised water standard solutions. For the river samples B and C and the saline samples, the ratio is highly consistent and not significantly different from 1.0. For the more highly coloured water (A), there is a marked suppressive interference which is more variable from run to run. This is probably attributable to coextraction of humic material which was clearly visible as both colour and solid precipitate in the alcohol layer. Consequently, data for Sample A have been reported after standard additions calibration. Data for the other samples were calculated by direct comparison with aqueous standards.
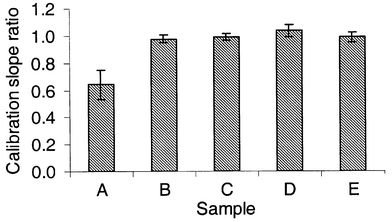 | ||
| Fig. 2 Ratio of calibration slope in the sample matrix versus that in deionised water standard solutions. Error bars are 95% confidence limits on the mean slope for 9 analytical runs. | ||
To assess the likelihood of losses of the determinand by adsorption during filtration, a river water sample was prefiltered, spiked with CrVI and analysed with and without filtration. The concentration in the filtered sample was found to be 1.33 ± 0.1 (p = 0.05)for the unfiltered sample and 1.34 ± 0.04 for the filtered sample. This indicates minimal loss of determinand by adsorption during filtration. The stabilty of CrVI in water samples has been demonstrated as at least one month by Sirinawin and Westerlund,16 so there does not appear to be a need to extract samples immediately.
Conclusions
This study shows how hexavalent chromium may be determined at sub- microgram per litre concentrations. This complexation/preconcentration procedure is capable of determining dissolved hexavalent chromium in fresh and saline samples in the range 0.03 to 2 μg l−1. The technique is adequately precise to be used for compliance monitoring to a proposed water quality criterion of 2 μg l−1. Recoveries of CrVI from a range of waters were not significantly outside the range 90–110%, though for samples high in dissolved organic carbon (>6 mg l−1 C), standard additions calibration is required. The technique is simple to use and requires instrumentation that is available in most laboratories.References
- H.E Allen, R. H. Hall and T. D. Brisbin, Environ. Sci. Technol., 1980, 14, 441–443 CAS.
- California Dept. of Health Services (1999). http://www.dhs.ca.gov/ps/ls/elap/html/chrom6.htm.
- K. Ishiki, Y. Sohrin, H. Karatani and E. Nakayama, Anal. Chim. Acta, 1989, 224, 55–64 CrossRef.
- R. E. Cranston and J. W. Murray, Anal. Chim. Acta, 1978, 99, 275–284 CrossRef CAS.
- M. J. Gardner and J. E. Ravenscroft, Fresenius' J. Anal. Chem., 1996, 354, 602–605 CAS.
- E. Beceiro-Gonzalez, J. Barcela-Garcia, P. Bermejo-Barrera and A. Bermejo-Barrera, Fresenius' J. Anal. Chem., 1992, 344, 301–305 CrossRef CAS.
- M. Boussemart, C. M. G. van den Berg and M. Ghaddaf, Anal. Chim. Acta, 1992, 262, 103–115 CrossRef CAS.
- D. M. Adria Cerezo, M. Llobat Estelles and A. R. Mauri Aucejo, Talanta, 2000, 15, 287–292.
- P. K. G. Manninen, Int. J. Environ. Anal. Chem., 1999, 75, 43–56 Search PubMed.
- Method 1636, Determination of Hexavalent Chromium by Ion Chromatography, US EPA, 1995, http://www.epa.gov/cgi-bin/claritgw Search PubMed.
- Standard Methods for the Examination of Water and Wastewater, American Public Health Association, Washington DC, 19th edn., 1995, ISBN 0–87553–223–3 Search PubMed.
- E. B. Sandell and H. Onishi, Photometric Determination of Traces of Metals, John Wiley, Chichester, 1978, 4th edn., pp. 1082, ISBN 0–471–03094–5 Search PubMed.
- J. Timmerman, M. J. Gardner and J. E. Ravenscroft, UN/ECE Task Force on Monitoring and Assessment, Volume 4—Quality Assurance, 1996, ISBN 9036945860 Search PubMed.
- R. V. Cheeseman, M. J. Gardner and A. L. Wilson, A Manual on Analytical Quality Control for the Water Industry, WRc Report NS30, 1989, WRc, Marlow, Bucks., ISBN 0902156853 Search PubMed.
- ISO/TR 13530 (1998) Water Quality—“Guide to Analytical Quality Control for Water Analysis”.
- W. Sirinawin and S. Westerlund, Anal. Chim. Acta, 1997, 356, 35–40 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |