Rapid determination of underivatized pyroglutamic acid, glutamic acid, glutamine and other relevant amino acids in fermentation media by LC-MS-MS
Jun
Qu
a,
Wei
Chen
a,
Guoan
Luo
*a,
Yiming
Wang
a,
Shengyuan
Xiao
a,
Zhihua
Ling
b and
Guoqiang
Chen
b
aAnalysis Center, School of Life Science and Engineering, Tsinghua University, Beijing, 100084, China. E-mail: galuo@chem.tsinghua.edu.cn; Fax: +86 10 62781688; Tel: +86 10 62781688
bDepartment of Biology, School of Life Science and Engineering, Tsinghua University, Beijing, 100084, China
First published on 11th December 2001
Abstract
Determination of amino acids in a complex matrix without derivatization is advantageous, however, difficulties are found in both the detection and the separation of those compounds. In this study, a rapid and reliable LC-MS-MS method for the quantitation of underivatized amino acids in exocellular media was established. Injections were made directly after centrifugation of the samples, without further preparation. The separation of seven underivatized amino acids was achieved on a reversed-phase C18 column with pentadecafluorooctanoic acid as a volatile ion-pair reagent, and the specific detection of most amino acids was achieved by MS-MS of the specific transitions [M + H]+→[M + H − 46]+. The calibration curves of all analytes were linear over the range of 1.0–1000 μg ml−1 and the detection limits ranged from 0.1 to 5 ng ml−1, with an injection volume of 20 μl. The inter-day and intra-day precisions ranged from 2.6 to 5.7% and 4.8 to 8.2%, respectively; the mean recoveries of the seven analytes were 81–104%, 91–107% and 93–101% respectively at the spiked level of 10, 40 and 200 μg ml−1. A large number of fermentation samples were analysed using this method. The technique is simple, rapid, selective and sensitive, and shows potential for the high-throughput quantitation of amino acids from other biological matrices.
Introduction
Glutamic acid (Glu) and glutamine (Gln) are important amino acids (AA) in the fields of medicine, food industry, biotechnology, biochemistry, etc.1 Since the 1950s they have been produced by large-scale bacterial fermentation; the current annual production of glutamate alone is approximately 900 000 t and increases each year.2 It has been reported that the strains for the production of Glu and Gln could also produce other AA or AA derivatives, primarily pyroglutamic acid (Pyro-Glu), N-acetylglutamine (N-Acetyl-Gln), threonine (Thr), proline (Pro) and valine (Val).3,4 This has led to extensive screening and breeding programs to develop strains for the production of these amino acids.5In order to screen suitable strains and to study the effect of various culture conditions on the production of certain AA, a high-throughput, sensitive, and accurate method for the determination of specific AA in fermentation media is needed. Ion exchange chromatography and high performance liquid chromatography (HPLC) are the most commonly applied methods for the determination of AA.6–9 These two methods have enabled qualitative and quantitative determination of most AA. However, both the methods require laborious sample preparation when assaying complex biological samples and derivatization (either pre- or post-column) must be employed, in most cases, to increase sensitivity and/or improve separation. In addition, the analysis times of these two methods are lengthy and some AA derivatives are unstable. Other analytical methodologies that have been applied to the determination of AA include gas chromatography (GC),6 thin-layer chromatography (TLC),6 capillary electrophoresis (CE),10,11 gas chromatography-mass spectrometry (GC-MS),12 liquid chromatography-mass spectrometry (LC-MS),13 tandem mass spectrometry (MS-MS)14 and liquid chromatography-tandem mass spectrometry (LC-MS-MS).15 Most current methods require derivatization (in most cases, either the derivatization procedures are time-consuming or the derivatives are not stable) and laborious sample preparation when assaying biological samples. Determination of AA extracted from biological matrices without derivatization is advantageous because it not only eliminates laborious sample preparation procedures but also reduces the errors introduced by those procedures.
The problems of assaying underivatized AA consist in both the detection and the separation of these compounds. There are reports on the determination of free AA by the addition of Cu(II) to the HPLC mobile phase, making the AA UV-detectable.16,17 This method did eliminate the derivatization procedure, however the present of Cu(II) in the mobile phase not only compromised chromatographic resolution but also could cause corrosion of the components of some HPLC system. Some methods for the determination of several underivatized AA by HPLC coupled with electrochemical detectors have also been reported.18,19 For most established HPLC-related methods, baseline separation of all analytes is needed; this results in relative long analysis times and limits their application in large-scale analysis. With the development of MS-MS methods, high sensitivity and selectivity can be achieved in the analysis of complex samples. This opens the possibility that trace amounts of AA could be determined without derivatization and baseline chromatographic separation. In a recent study,20 a mixture of underivatized AA standards was investigated by LC-MS. However, the quantitation of underivatized amino acids in biological matrices by LC-MS-MS has not been reported.
This paper proposes a LC-MS-MS method for the quantitation of free AA in exocellular samples with high speed, sensitivity and selectivity and without the need for derivatization. Volatile mobile phase modifiers were used to enable separation of AA on a reversed-phase column and specific transitions were employed for the MS-MS detection to ensure the selectivity of this method. Glu, Gln, Pyro-Glu, N-Acetyl-Gln, Thr, Pro and Val were determined simultaneously in the fermentation media of some strains of Corynebacterium.
Experimental
Chemicals
Pyro-Glu, N-Acetyl-Gln and other a-AA were purchased from Sigma Chemical Company (St. Louis, MO, USA). Stable isotopes of AA (as internal standards) including Val (D8, 98%), Thr (13C2, 98%), Glu (D3, 98%), Gln (D5, 98%) and Pro (D7, 98%) were from Cambridge Isotopes (Andover, MA, USA). HPLC grade acetonitrile (ACN) and methanol were from Tedia Inc. (Fairfield, OH, USA). Formic acid (88%) and trifluoroacetic acid (TFA, 98%) was obtained from Fluka (Buchs, Switzerland) and pentadecafluorooctanoic acid (PDFOA, 98%) was from Acros Organics (Pittsburgh, PA, USA).Microorganisms
The strains of Corynebacterium used for this study, such as Corynebacterium glutamicum (ATCC 14067 and ATCC 14752) and Corynebacterium acetoacidophilum (ATCC 13870) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA).Sample preparation
A 20 μl portion of fermentation media was transferred to a 2 ml vial and 980 μl of methanol which contained 50 μg ml−1 of each internal standard and 0.5% formic acid was added to the vial. After vortex-mixing for 10 min and centrifugation at 5000g for another10 min, the supernatant was ready for injection.LC-MS-MS conditions
A PE SCIEX (Toronto, Canada) API 3000 triple-quadrupole tandem mass spectrometer equipped with a Turbo Ion Spray Interface, an online degasser and a Perkin-Elmer binary pump (model 250) was used for LC-MS-MS analysis. For the investigation of the product spectra of AA, 10 μg ml−1 standard solutions were directly infused to the interface by a syringe pump at a rate of 0.3 ml h−1. Separation of AA was carried out on a Dikama Diamonsil C18 (particle size 5 μm, 150 × 2.1 mm) column maintained at 40 °C. The mobile phase consisted of two solvents: 1.0 mM PDFOA and 0.05% TFA in water (solvent A, pH = 2.8), and acetonitrile (solvent B). Gradient conditions: the proportion of B was 5% for the first 4 min; then increased to 45% over 3 min; then 45% B was maintained till the end of analysis. The flow rate was 0.25 ml min−1 and the injection volume was 20 μl. The LC effluent was split 4∶1 using a post column split before the interface. Multi reaction monitoring (MRM) of MS-MS was used for the specific detection of AA and their internal standards. The dwell time for each transition was 75 ms and the pause time for the changes of scan parameter was 8 ms. The pressure of the collision gas (N2) was 2.4 mTorr and corresponding collision energies were applied respectively for each transition. The flow rates of nebulizer gas (air), curtain gas (N2) and drying gas (N2) were respectively 10 ml min−1, 12 ml min−1, 1.2 l min−1. The ion spray voltage, orifice potential and ring focus voltage were set at 4800 V, 25 V and 160 V, respectively.Calibration and method validation
Deuterated or 13C-labelled internal standards were used for calibration. Standard solutions of AA were prepared at the concentration of 0.5, 2.0, 10, 50, 250 and 1000 μg ml−1 in a solvent consisting of methanol–water–formic acid 80∶20∶0.1 (v/v/v) and 50 μg ml−1 of each internal standard. The data were processed using Macquan software (PE Sciex): calibration curves were prepared by plotting analyte–internal standard extracting ion current (XIC) peak area ratios vs. concentrations; determination of AA from fermentation samples was performed using weighted least-squares regression analysis of the standard curves.The recoveries of AA from fermentation media were determined by spiking AA to a fermentation media sample at three levels (10, 40 and 200 μg ml−1), then experimentally measuring the added amounts. Precision of the assay was calculated by repeat analysis of the same sample and was estimated as the RSD (%) of the replicate measurements, respectively, intra-day and inter-day.
Results and discussion
LC-MS-MS optimization
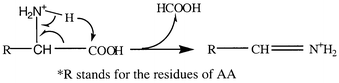
Since the molecular weights of the AA studied are below 200, the interferences from solvents and sample matrix are severe in the mass spectrum. Specific MS-MS transitions as well as HPLC separation are therefore necessary for the development of a satisfactory quantitative method. In order to select an optimal transition for the MS-MS detection of an analyte, two factors should be considered: first, the transition should be compound-specific; second, the intensity of the product ion should be sufficiently abundant for detection. We investigated the product spectra of the seven analytes under positive ion mode, and the collisionally activated dissociation (CAD) product ions of those AA are shown in Table 1. The product ions of each AA are sorted by their maximum intensities (i.e. the intensity of a product ion acquired under its optimal MS parameters, especially the collision energy), from abundant to weak. Most AA have an abundant product at [M + H − 46]+ among their CAD fragments, which corresponds to the neutral loss of formic acid by a rearrangement:This transition is specific to α-AA because it involves both the carboxyl and α-amino groups. We therefore selected this transition for the detection of most AA. For Gln, the product [M + H − 46]+ was too weak to be used for a sensitive detection, therefore the product [M + H − NH3]+ (m/z 130) was chosen for MRM. The transitions and their corresponding optimal collision energies chosen for MRM of AA are listed in Table 2.
| Compound | [M + H]+ | Product ions (sorted by maximum ion intensities, from abundant to weak) |
|---|---|---|
| a Denotes the product [M + H − 46]+. | ||
| Val | 118 | 72a, 55 |
| Thr | 120 | 74a, 102, 56, 84, 88, 92 |
| Glu | 148 | 84, 102a, 130, 56, 41 |
| Gln | 147 | 130, 84, 56, 101a, 47 |
| Pro | 116 | 70a, 60, 57 |
| Pyro-Glu | 130 | 84a, 77, 102 |
| N-Acyl-Gln | 189 | 143a, 172, 84, 47 |
| AA | Transitions (AA/internal standard) | Collision energy/eV | Retention time/min | Detection limit/ng ml−1 |
|---|---|---|---|---|
| Val | 118→72/126→80 | 15 | 9.3 | 0.1 |
| Thr | 120→74/122→76 | 15 | 4.5 | 1 |
| Glu | 148→102/151→105 | 15 | 3.3 | 5 |
| Gln | 147→130/152→135 | 13 | 4.0 | 2 |
| Pro | 116→70/123→77 | 21 | 6.2 | 1 |
| Pyro-Glu | 130→84/152→135 | 15 | 2.3 | 0.5 |
| N-Acyl-Gln | 189→143/152→135 | 17 | 4.4 | 4 |
Even with specific MS-MS detection, HPLC separation was essential in order to eliminate interferences from sample matrix and from other analytes. Reversed-phase chromatography is normally not applicable for the determination of underivatized AA because these compounds lack larger hydrophobic side-chains. Therefore, mobile phase modifiers should be used to improve separation of AA. In order to improve separation of AA on a reversed-phase column and at the same time, avoid compromising ionization efficiency in the turbo ion spray interface, we adopted an acetonitrile–water gradient, using a combination of PDFOA and TFA (both are volatile) as modifiers in the mobile phase. Additional functions of TFA in the mobile phase included increasing the MS signal of AA, improving peak shape and speeding up the elution of some AA. Under these HPLC conditions, good LC-MS-MS chromatographic separation was achieved in the assay of fermentation samples. This was exemplified by the successful discrimination of Pyro-Glu from Glu and Gln, etc. A typical chromatogram of the analysis of fermentation media by LC-MS-MS is shown in Fig. 1. Retention times and detection limits (S/N = 3) for the seven analytes are shown in Table 2.
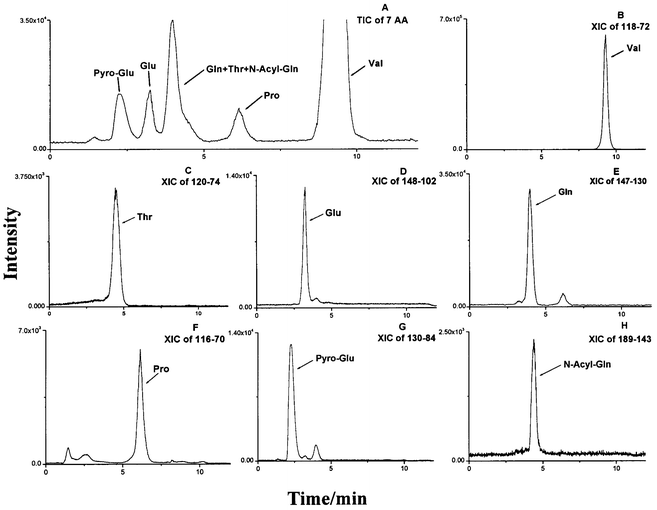 | ||
| Fig. 1 The chromatograms of LC-MS-MS quantitation of seven AA from fermentation media of Corynebacterium acetoacidophilum (ATCC 13870). (A) TIC (total ion current); (B)–(H) XIC (extracting ion current) chromatograms respectively of seven AA. The cells were cultivated in a medium containing glucose, 200 g l−1; NH4Cl, 50 g l−1; KH2PO4, 0.7 g l−1; MgSO4·7H2O, 0.4 g l−1, biotin, 0.3 g l−1; etc; the culture was carried out at 32 °C, on a rotary shaker for 48 h. | ||
Problems associated with the use of PDFOA
It was observed that the retention times of AA increased slightly when a number of analyses were performed. The retention times of some AA increased by about 1 min after 12 consecutive assays. As a consequence, the quantitation was rejected by Macquan software because of peak migration. This effect occurred because PDFOA accumulated in the column and slowly modified the surface of stationary phase. To avoid this, we flushed the column with 100% ACN for 20 min to remove the accumulated PDFOA after every six chromatographic runs.Quantitation of AA in fermentation media
The calibration curves showed good linearity over the concentration range of 1.0–1000 μg ml−1 (r2 = 0.987–0.999, shown in Table 3). Besides Gln, the D5-Gln also served as the internal standards for Pyro-Glu and N-Acetyl-Gln. The analytical recoveries, intra- and inter-day precisions for the determination of AA from fermentation media of Corynebacterium glutamium (ATCC 14067) are also shown in Table 3. The quantitative data for the determination of AA in other strains are analogous to those of ATCC 14067 (data not shown). Matrix effects that could severely impair the LC-MS-MS quantitation21,22 of biological samples were not observed, as demonstrated by the good recoveries and precisions of the analyses. This is attributable to the use of stable isotopes as internal standards and the adequacy of the HPLC separation. The analysis time was 12 min per sample, suitable for large-scale analysis.| AA | Linearity of calibration (r2) | Recovery (s; %)a | Intra-day precision RSD (%)b | Inter-day precision RSD (%) |
|---|---|---|---|---|
| a The recoveries were determined in triplicate, respectively, at concentrations of 10, 40 and 200 μg ml−1. b Aliquots of fermentation media sample stored at −20 °C were analysed six consecutive times in one day (intra-day, n = 6), and twice on three different days (inter-day, n = 6). | ||||
| Val | 0.999 | 99 (5), 103 (8), 97 (6) | 5.7 | 7.4 |
| Thr | 0.998 | 101 (6), 95 (4), 100 (8) | 6.2 | 5.7 |
| Glu | 0.995 | 104 (3), 97 (2), 99 (3) | 2.6 | 5.9 |
| Gln | 0.994 | 94 (6), 103 (7), 101 (3) | 3.6 | 8.2 |
| Pro | 0.999 | 97 (5), 107 (4), 100 (3) | 3.4 | 5.9 |
| Pyro-Glu | 0.991 | 93 (2), 91 (5), 93 (3) | 4.2 | 4.8 |
| N-Acyl-Gln | 0.987 | 89 (5), 93 (2), 95 (4) | 2.9 | 6.3 |
In our study of the production of AA by biotechnological methods, about 300 fermentation samples have been assayed by this method. Some of those samples were also assayed by an established HPLC method using an automated on-line HPLC system with pre-column derivatization (o-phthaldialdehyde–3-mercaptopropionic acid) and with norvaline as the internal standard. The quantitative results by the two methods were similar (data not shown). However, the mean recovery values for AA by the HPLC method were significantly lower than those found using the LC-MS-MS method. We believe that this is a consequence of both the inefficiency of the derivatization and the use of only one or two internal standards for the quantitation of the seven AA, most of which possess different chemical and physical properties (such as pI value, solubility, etc.) from those of the internal standard.
The LC-MS-MS method can also be used for rapid quantitation of AA in other biological matrices, especially when both accuracy and speed are required for the assay.
Acknowledgement
This work was financially supported by the Chinese National Key Foundation of Science (Grant No. 1999054404).References
- J. D. Kopple, in Amino Acids, Metabolism and Medical Applications, ed. G. L. Blackburn, J. P. Grant and V. R. Young, John Wright, Boston, 1983, pp. 451–471 Search PubMed.
- S. Delaunay, P. Gourdon, P. Lapujade, E. Mailly, E. Oriol, J. M. Engasser, N. D. Lindley and J.-L. Goergen, Enzyme Microb. Technol., 1999, 25, 762 CrossRef CAS.
- C. A. Batt, M. T. Follettie, H. K. Shen, P. Yek and A. J. Sinskey, Trends Biotechnol., 1985, 3, 305 CrossRef CAS.
- T. Nakanishi, J. Ferment. Technol., 1978, 56, 179 Search PubMed.
- M. Nampoothiri and A. Pandey, Proc. Biochem., 1998, 33, 147 Search PubMed.
- G. W. Robinson, in Amino Acid Determination Methods and Techniques, ed. S. Blackburn, Marcel Dekker, New York, 1978, pp. 39–85 Search PubMed.
- D. W. Hill, F. H. Walters, T. D. Wilson and J. D. Stuart, Anal. Chem., 1979, 51, 1338 CrossRef CAS.
- E. B. A. Mohammed and A. G. S. Mohammed, Analyst, 1997, 122, 147 RSC.
- J. You, W. Lao, J. You and G. Wang, Analyst, 1999, 124, 1755 RSC.
- Y. H. Lee and T. I. Lin, J. Chromatogr. A, 1994, 680, 287 CrossRef.
- O. Shigeyuki, F. Toshiaki and M. Yasuyoshi, Analyst, 1996, 121, 1683 RSC.
- C. D. Marquez, S. T. Weintraub and P. C. Smith, J. Chromatogr., B: Biomed. Appl., 1994, 658, 213 Search PubMed.
- K. Fujii, Y. Ikai, T. Mayumi, H. Oka, M. Suzuki and K. Harada, Anal. Chem., 1997, 69, 3346 CrossRef CAS.
- D. H. Chance, D. S. Millington, N. Terada, S. G. Kahler, C. R. Roe and L. F. Hofman, Clin. Chem., 1993, 39, 66 Search PubMed.
- J. Qu, Z. Wu, G. Luo, M. Liu and C. Yang, Chem. J. Chin. Univ., 2000, 21, 210 Search PubMed.
- S. Levin and E. Grushka, Anal. Chem., 1985, 57, 1830 CrossRef CAS.
- B. Polanuer, A. Sholin, N. Dmina and N. Rumiantseva, J. Chromatogr., 1992, 594, 173 CrossRef CAS.
- K. Sato, J. Y. Jin, T. Takeuchi, T. Miwa, Y. Takekoshi, S. Kanno and S. Kawase, Analyst, 2000, 125, 1041 RSC.
- J. S. Soblosky, L. L. Colgin, C. M. Parrish, J. F. Davidson and M. E. Carey, J. Chromatogr., B: Biomed. Appl., 1998, 712, 31 Search PubMed.
- K. Petritis, P. Chaimbault, C. Elfakir and M. Dreux, J. Chromatogr. A, 2000, 896, 253 CrossRef CAS.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 1998, 70, 882 CrossRef CAS.
- J. Qu, Y. Wang and G. Luo, J. Chromatogr. A, 2001, 919, 437 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |