Determination of an arsenosugar in oyster extracts by liquid chromatography-electrospray mass spectrometry and liquid chromatography-ultraviolet photo-oxidation-hydride generation atomic fluorescence spectrometry
D. Sánchez-Rodas*a, A. Geiszingerb, J. L. Gómez-Arizaa and K. A. Francesconib
aDepartamento de Química y Ciencia de los Materiales, Escuela Politécnica Superior, Universidad de Huelva, E-21819 Palos de la Frontera, Huelva, Spain. E-mail: rodas@uhu.es; Fax: +34 959 017414; Tel: +34 959 017440
bInstitute of Biology, University of Southern Denmark, DK-5230 Odense M, Denmark
First published on 13th December 2001
Abstract
HPLC-UV-HG-AFS analysis of aqueous extracts of oysters (Crassostrea gigas) taken from the southwestern Atlantic coast of Spain showed the presence of arsenite, arsenate, dimethylarsinic acid and an unidentified arsenic peak. Subsequent analysis of the oyster samples by LC-electrospray MS and comparison with four standard dimethylarsinoylribosides (arsenosugars), showed that the previously unidentified peak was an arsenosugar (arsenosugar 2). When the arsenosugar in the oyster was quantified using the two detection methods and external calibration with standard arsenosugar, there was a large discrepancy between the two sets of results. The LC-MS analysis was strongly affected by the sample matrix and gave concentrations 50% lower than those obtained by AFS detection. When the method of standard addition was applied to the LC-MS analysis, the results were comparable to the AFS data. The matrix effects were eliminated by subjecting the extract to a clean-up procedure with anion-exchange and gel permeation preparative chromatography before the LC-MS analysis. The arsenosugars gave a small signal without photo-oxidation when they were analysed by HPLC-HG-AFS. Possibly this resulted from partial decomposition of the arsenosugar to dimethylarsinic acid under the acidic conditions employed in the hydride generation step.
Introduction
The biochemistry and biological fate of arsenic are of great interest, because of the wide range of arsenic compounds that occur in the environment and the different toxicities that they display.1 For reasons that are yet to be elucidated, this diversity of arsenic compounds is most apparent in biological samples of marine origin and about 25 such compounds have been reported so far.2 In most marine animals, the major arsenic compound is arsenobetaine, whereas marine algae contain a group of related arsenic-containing carbohydrates (dimethylarsinoylribosides) commonly referred to as arsenosugars.3 Arsenosugars also occur in marine animals, presumably as a consequence of food chains based on algae.A previous study4 with HPLC-UV-HG-AFS on the arsenic compounds in oysters (Crassostrea gigas) from the Spanish southwestern coast identified arsenite, arsenate, arsenobetaine and dimethylarsinic acid. A major arsenic compound in the oyster extracts, however, could not be identified by that technique. We report here the identification of this compound by LC-electrospray (ES) MS and its subsequent determination with HPLC-UV-HG-AFS. The results from the two methods are compared and the significant influence of the sample matrix on the quantification of the arsenosugar by LC-MS is discussed.
Experimental
Sampling area
Oysters (Crassostrea gigas) were collected in August 2000 along the common estuary formed at the mouth of the Tinto and Odiel rivers, on the Atlantic southwestern coast of Spain. This area is polluted by heavy metals, because both rivers drain a major mining region and there are also several processing plants along the estuary.5 The oysters grow on the rocks of an artificial barrier running for 15 km parallel to the coast, which serves to protect the entrance of the harbour of Huelva. Oysters were found along the last 9 km of the barrier. Four samples were taken from the inner side of the barrier, at 2 km intervals, and a fifth sample was taken from the outer side of the barrier, which faces open sea. The oysters (about 1 kg from each sampling point) were transported live to the nearby laboratory where the shells were removed and the whole fresh tissues (ca. 100 g) were freeze-dried (ca. 10 g dry mass), ground to a homogeneous powder and stored at −20 °C.Standards and reagents
Standard solutions of arsenic compounds were prepared from arsenic trioxide (Panreac, Barcelona, Spain), sodium arsenate (Na2HAsO4·7H2O) (Merck, Darmstadt, Germany), sodium methylarsonate (Carlo Erba, Milan, Italy) and dimethylarsinic acid (Sigma, St. Louis, MO, USA). Arsenosugars 2–4 (Fig. 1) were previously isolated from natural sources6 and arsenosugar 1 was synthesised by the method of McAdam et al.7 Additional quantities of arsenosugars 3 and 4 were obtained from the brown alga Fucus serratus using the preparative chromatographic techniques (anion exchange and gel permeation) described in earlier studies.6 Chemicals used in HPLC mobile phases (KH2PO4, K2HPO4, NH4HCO3, pyridine and methanol) and HCl and NaOH employed for hydride generation were all supplied by Merck, NaBH4 was obtained from Panreac and K2S2O8 (>99%) used in the photo-oxidation reaction was purchased from Aldrich (Steinheim, Germany).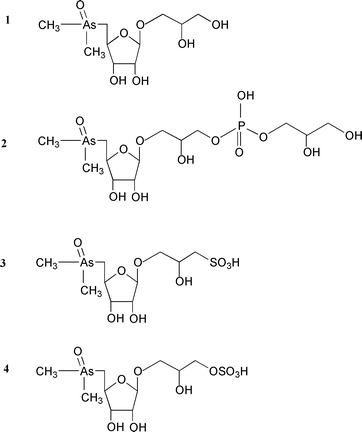 | ||
| Fig. 1 Structures of the four major arsenosugars occurring in marine samples. | ||
HPLC-UV-HG-AFS
The arsenic compounds were separated and detected with instrumentation consisting of a liquid chromatograph, on-line UV photo-oxidation, hydride generation and atomic fluorescence detection.8 The liquid chromatograph consisted of a JASCO PU 1580 HPLC pump (binary pump with a solvent degasser), equipped with a PRP X-100 anion-exchange column (250 × 4.1 mm id, 10 μm particle size) from Hamilton (Reno, NV, USA) equilibrated with 10 mM buffer (KH2PO4–K2HPO4) (pH 5.8), at a flow rate of 1.1 mL min−1. Hydride generation was achieved by adding solutions of 1.5 M HCl and NaBH4 (1.5% w/v in 1% NaOH ), each at 1 mL min−1 with the aid of a peristaltic pump (Minipuls 3, Gilson, Villiers-le-Bel, France). On-line photo-oxidation was carried out by adding a basic solution of potassium persulfate (2% w/v K2S2O8 in 2% w/v NaOH) to the reaction coil (8 m) wrapped around a 15 W low pressure Hg lamp (TNN 15/32, Heraeus, Germany). When photo-oxidation was employed, the HCl concentration was increased from 1.5 to 8 M. The volatile hydrides were transported to a gas–liquid separator, using argon as carrier gas. An Excalibur 10.33 instrument (PS Analytical, Orpington, Kent, UK) was used for atomic fluorescence spectrometric (AFS) detection, employing as radiation source a boosted-discharge hollow-cathode lamp (Photron, Victoria, Australia). Hydrogen (60 mL min−1) was added at the gas–liquid separator to support the hydrogen–argon diffusion flame of the detector.LC-MS
The separation and detection of the arsenosugars were carried out using a Hewlett-Packard LC-MS system, consisting of a 1100 Series high-performance liquid chromatograph (solvent degasser, binary pump, autosampler and thermostatted column compartment) and a G1956A MSD single-quadrupole mass spectrometer equipped with an atmospheric pressure ionization (API) interface. The optimum conditions of operation of the MS for determining arsenosugars have been described previously.9 The following m/z values were selected for the identification of the protonated molecules [M + H]+ of the arsenosugars: arsenosugar 1, m/z 329; arsenosugar 2, m/z 483; arsenosugar 3, m/z 393; and arsenosugar 4, m/z 409. Anion-exchange chromatography was performed using a PRP X-100 column (150 × 4.1 mm id, 10 μm particle size) from Hamiton, thermostatted at 30 °C. The mobile phase was a mixture of 20 mM NH4HCO3 at pH 9.5 (adjusted with aqueous NH3) and methanol (7 + 3 v/v) at a flow rate of 1 mL min−1. Cation-exchange chromatography was performed using an Ionospher C column (100 × 3 mm id, 5 μm film thickness) from Chrompack (Hundige, Denmark), thermostatted at 30 °C. The mobile phase was a mixture of 20 mM pyridine at pH 2.6 (adjusted with aqueous HCOOH) and methanol (8 + 2 v/v) at a flow rate of 1 mL min−1. Quantification was performed by both external calibration and the method of standard additions. In the latter case, 10 μL of water or 10 μL of the arsenosugar standard (50, 100 or 150 μg As L−1) were added to 10 μL aliquots of the aqueous extract.Total arsenic determinations
Total arsenic concentrations in the freeze-dried oyster samples, the aqueous extracts and the solid residue after extraction (pellet) were determined by hydride generation atomic absorption spectrometry (Perkin-Elmer MHS 20 mercury–hydride system and a Perkin-Elmer 2380 atomic absorption spectrometer) following mineralisation in a muffle furnace after adding a slurry consisting of Mg(NO3)2 and MgO.10The accuracy of the method was checked by analysing the certified reference material DORM-2 dogfish muscle (National Research Council of Canada, Ottawa, Canada), with a certified value of 18.0 ± 1.1 mg As kg−1, (found 17.8 ± 0.7 mg As kg−1, n = 4).Extraction of arsenic species
The freeze-dried oyster powder (250 mg) was weighed and transferred into a polyethylene test-tube. Distilled water (5.00 mL) was added and the samples were extracted with a sonication probe (2 × 30 s). The resulting homogenate was centrifuged (40000 rpm, 20 min) and the supernatant filtered (0.45 μm); portions of these filtrates were injected directly on to the chromatographic columns. The solid residue (pellet) remaining after centrifugation was kept for total arsenic determination.Purification of oyster extracts
The freeze-dried oyster sample (2.00 g) was extracted with methanol (3 × 20 mL), centrifuged (15000 rpm for 15 min) and the supernatant was evaporated to dryness under reduced pressure (Büchi rotavapor, 40 °C). The residue was redissolved in water (3 mL), filtered (0.45 μm) and the filtrate applied to a gel permeation column (Sephadex G-15, Pharmacia Biotech, 700 × 26 mm id), using water (0.3 mL min−1) as eluent. Fractions (5 mL) were collected and arsenic was determined by electrothermal (ET) AAS (Zeeman 3030, Perkin-Elmer). The arsenic containing fractions were combined, evaporated to a small volume (3 mL) and applied to an anion-exchange column (DEAE Sephadex A-25, 200 × 16 mm id). Elution was performed with 0.05 M TRIS buffer (pH 8.0, adjusted with HCl) at a flow rate of 1 mL min−1. Fractions (5 mL) were collected and analysed for arsenic by ETAAS; those fractions corresponding to arsenosugar 2 were combined and evaporated to a volume of about 4 mL. The arsenosugar was then separated from the TRIS buffer by passing this solution through a gel permeation column (Sephadex G-15, 700 × 26 mm id, eluent water).Results and discussion
Total arsenic in oysters and oyster extracts
Total arsenic contents were determined in the freeze-dried samples before extraction and in the supernatant and the pellet obtained from centrifugation of the extracted sample. The four oyster samples taken inside the harbour contained similar arsenic concentrations (17.4–19.1 μg As g−1), all of which were higher than the sample taken from outside the harbour (12.5 μg As g−1). Although this may indicate a source of arsenic contamination inside the harbour, more samples would be needed to establish if this was in fact the case. Most of the arsenic in the oysters was water soluble: the extraction efficiencies, from a single aqueous extraction, ranged from 62 to 70% (Table 1). Previous work has shown that water is an efficient extractant of most arsenic compounds found in marine organisms.11,12 The arsenic left unextracted by our method is possibly lipid As or protein-bound As. The recovery of arsenic [(Assupernatant + Aspellet)/Asinitial freeze-dried sample] was 94–105% for the five oyster samples.Examination of arsenic species by HPLC-(UV)-HG-AFS
Five arsenic standards [As(III), As(V), AsB, DMA and MMA] were analysed using AFS detection, without and with the photo-oxidation step [Fig. 2(a) and (c)]. The oyster extracts were shown to contain As(III), AsB, DMA and As(V) by comparison of the retention times of arsenic standards and sample compounds run without and with a photo-oxidation step [Fig. 2(b) and (d)]. We note that the identification of AsB is not definitive because other refractory cationic arsenicals (e.g., tetramethylarsonium ion) may behave similarly under these conditions. In addition, the oyster samples contained a large As signal [with photo-oxidation, Fig. 2(d)] at a retention time (6.9 min) which did not match any of the arsenic standards available at that time. These data were similar to previously reported results.4An interesting characteristic of the unidentified compound was that it gave a small signal without photo-oxidation [Fig. 2(b)] immediately after acidification. This suggested that the compound might be reduced to an arsine, albeit a relatively involatile one, or that the acidic conditions were sufficient to decompose the original arsenical partly to one that could produce a volatile arsine. Alternatively, the signal may have resulted from two arsenic compounds with the same retention times, only one of which gave a volatile arsine without photo-oxidation.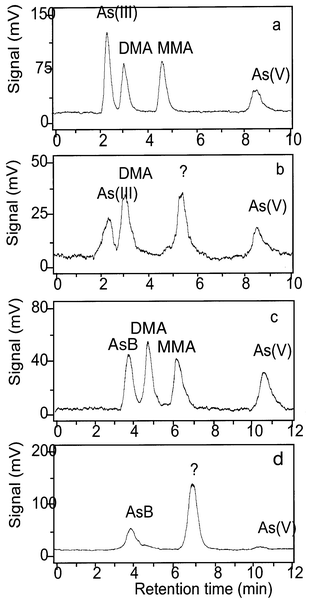 | ||
| Fig. 2 HPLC-(UV)-HG-AFS without on-line photo-oxidation (a and b) and with photo-oxidation (c and d): 25 μg As L−1 standards (a and c), oyster 2 extract (b and d). Chromatographic conditions: PRP-X100 column; 10 mM K2HPO4–KH2PO4 (pH 5.8), 1.1 mL min−1. Note that the retention times are about 1.5 min longer when the photo-oxidation step is included because the components must pass through an 8 m long reaction coil. | ||
Identification of the unknown arsenic compound by LC-ES-MS
The chromatographic properties of the unidentified arsenic species in the oyster extracts were consistent with its being an arsenosugar. Arsenosugars have been commonly reported in bivalve molluscs13 and they might be expected partly to degrade in acid to DMA,14 which would explain the observed AFS data. The crude oyster extracts were then analysed by LC-ES-MS to test the presence of the four arsenosugars (see Fig. 1) commonly found in marine samples. The technique has been used previously9,12 to determine arsenosugars in crude extracts of brown algae and found to be relatively free of interferences. Chromatograms for the four arsenosugar standards are shown in Fig. 3. When the technique was applied to the oyster extract, a signal with m/z 483 was observed which corresponded to the protonated molecular species [M + H]+ for arsenosugar 2 [Fig. 4(a)]. There were no corresponding [M + H]+ signals for the arsenosugars 3 or 4. There was also no signal for arsenosugar 1, although the detection of this compound may have been compromised because it elutes near the void volume. When the oyster extract was analysed on the cation-exchange column, an [M + H]+ signal was obtained matching the retention time of arsenosugar 1, but the identification could not be confirmed with detection of product ions (see below).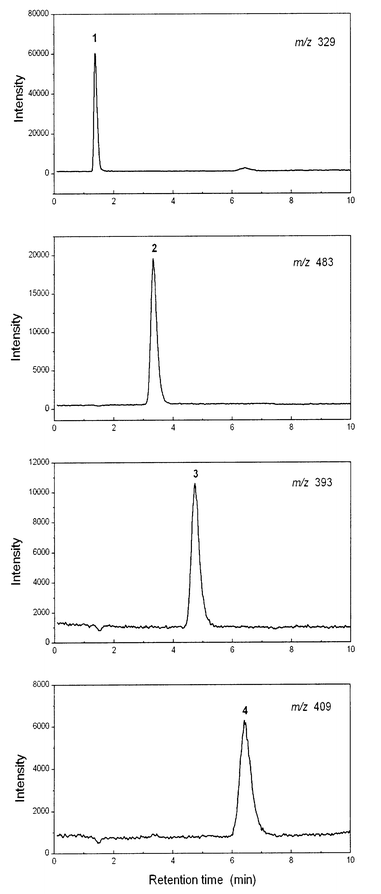 | ||
| Fig. 3 LC-ES-MS of 100 μg As L−1 arsenosugar standards, at the m/z of the protonated molecular species. Chromatographic conditions: PRP-X100 column; 20 mM NH4HCO3 (pH 9.5)–methanol (7 + 3), 1.0 mL min−1. | ||
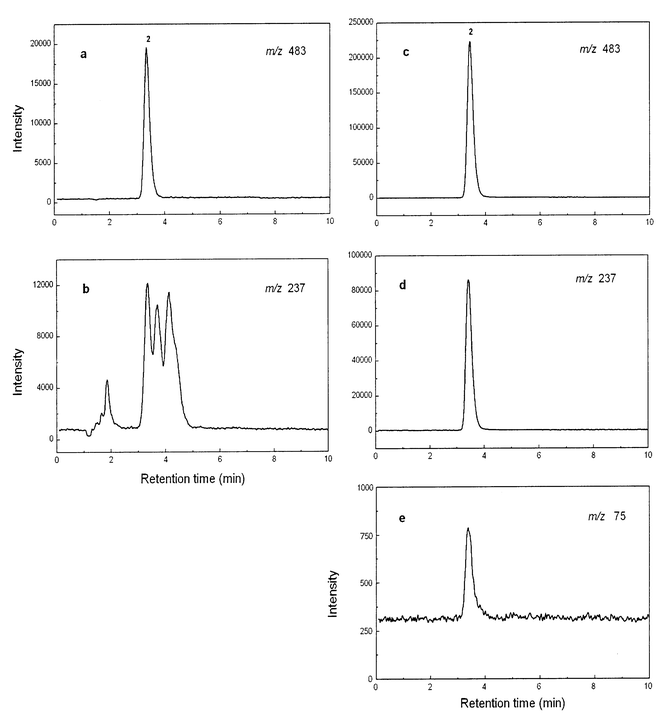 | ||
| Fig. 4 LC-ES-MS of oyster 2 extracts at different m/z values: before clean-up (a and b) and after clean-up (c, d and e). Chromatographic conditions as in Fig. 3. | ||
A valuable feature of arsenic speciation analysis with LC-ES-MS is that it can simultaneously provide both elemental and molecular information about the arsenic species.9 For arsenosugar 2, coincidence of retention times between standard and sample signals for m/z 75 (As+), m/z 237 (product ion characteristic of arsenosugars) and m/z 483 ([M + H]+) would provide strong evidence for the presence of the compound in the oyster extracts. However, although the m/z 483 detection [Fig. 4(a)] gave a clean trace with only one signal (which matched the retention time for the standard), the m/z 237 detection [Fig. 4(b)] produced several peaks that were not resolved from the likely product ion (retention time 3.4 min). Furthermore, we could not obtain signals for m/z 75 (As) at the time of these analyses, a problem thought to be related to the crude sample matrix. To overcome problems associated with the sample matrix, we partially purified arsenosugar 2 in the oyster extract, using preparative anion-exchange and gel permeation chromatography and then repeated the LC-ES-MS analyses. On this occasion, all three signals [Fig. 4(c)–(e)] characteristic of arsenosugar 2 were obtained with retention times exactly matching those for the standard compound.
Quantification of arsenosugar 2 in oyster extract by LC-MS
Arsenosugar 2 was quantified in the crude oyster extract based on the signal for the protonated molecular species ([M + H]+, m/z 483). Previous work9,12 had shown that this signal provided the most reliable quantification data and that, for crude algal extracts, the sample matrix had only small effects on the signal intensity.12 Those extracts, however, were much more concentrated in arsenosugars (about 10–40 fold) than the oyster extract in the current study. Analysis of the crude oyster extract by the method of standard additions showed that there was a 50% reduction in signal intensity because of sample matrix effects. Quantification of arsenosugar 2 by the standard additions method gave a value of 3.1 mg As kg−1 in the initial freeze-dried oyster sample, compared with 1.5 mg As kg−1 by external calibration. When an oyster extract was subjected to a purification procedure involving preparative gel permeation and anion-exchange chromatography, the matrix effects were eliminated and there was no significant difference between the results obtained by external calibration and the method of standard additions.Determination of arsenosugars by HPLC-UV-HG-AFS
During the course of this study, four arsenosugar standards (Fig. 1) became available for use with the HPLC-UV-HG-AFS method. The retention time for the standard arsenosugar 2 matched the retention time of the unknown peak (6.9 min) in the oyster extracts [Fig. 5(a)]. No matrix effect was observed on the signal, so consequently the quantification of this arsenic species was performed by external calibration. It should be noted that when the four arsenosugars were analysed without the photo-oxidation step [Fig.5(b)], small signals were also obtained. Possibly, these arise from partial degradation of the arsenosugars to DMA under the acidic conditions used to produce the volatile arsines.14 Although no matrix effect was observed for the arsenosugar 2 in the sample, the signal resulting from arsenobetaine was markedly depressed by the matrix. These effects diminished as the sample was diluted. Presumably the fact that arsenobetaine eluted near the void volume (together with many other sample components) contributed to these effects. Additionally, because arsenobetaine is chemically very stable, it is decomposed to arsenate much less readily than are the arsenosugars.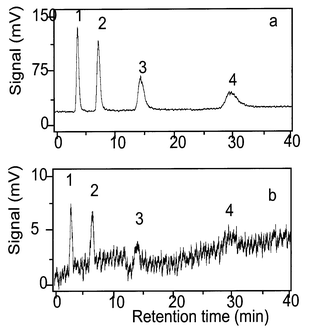 | ||
| Fig. 5 HPLC-UV-HG-AFS of 100 μg As L−1 arsenosugar standards: (a) with photo-oxidation and (b) without photo-oxidation. Chromatographic conditions as in Fig. 2. | ||
The concentration of arsenosugar 2 in the oyster sample (oyster 2, Table 1) obtained by AFS was 3.3 mg As kg−1, which agreed well with the value (3.1 mg As kg−1) obtained by LC-MS. Determination of arsenosugar 2 in the other oyster samples showed that it ranged from 12 to 19% of the total arsenic content (Table 1). Larsen15 reported that the arsenosugar content (sum of arsenosugars 1 and 2) in the NIST standard reference material (SRM) 1566a (Oyster Tissue) was ca. 18% of the total arsenic. Shibata and Morita16 reported arsenosugars 1 and 2 as major arsenic species in NIST SRM 1566 (Oyster Tissue). In scallops from Newfoundland, arsenosugars 1–4 were the major water-soluble arsenic compounds in the gonads, whereas only arsenosugar 4 was detected in muscle tissue.13 Arsenosugars 1 and 2 were also major arsenic compounds in bivalves from a hydrothermal vent in the Mid-Atlantic Ridge.17
Conclusions
The determination of arsenosugars in crude oyster extracts by LC-ES-MS can be influenced by the sample matrix. These effects can be overcome by incorporating sample clean-up steps prior to the LC-ES-MS analysis. HPLC-UV-HG-AFS is an alternative method for detecting arsenosugars in crude extracts since the signal response is little influenced by the sample matrix.Acknowledgements
We thank the Danish Research Council and the Royal Society of Chemistry (Daniel Sánchez-Rodas) for financial support under grant 0006/248 for international authors.References
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713 CrossRef.
- K. A. Francesoni and J. S. Edmonds, Adv. Inorg. Chem., 1997, 44, 147 Search PubMed.
- K. A. Francesconi and D. Kuehnelt, in Environmental Chemistry of Arsenic, ed. W. T. Frankenberger, Marcel Dekker, New York, in the press Search PubMed.
- J. L. Gómez-Ariza, D. Sánchez-Rodas, R. Beltrán and I. Giráldez, Int. J. Environ. Anal. Chem., 1999, 74, 203 Search PubMed.
- J. L. Gómez-Ariza, I. Giráldez, D. Sánchez-Rodas and E. Morales, Anal. Chim. Acta, 1999, 399, 295 CrossRef CAS.
- K. A. Francesconi, J. S. Edmonds, R. V. Stick, B. W. Skelton and A. H. White, J. Chem. Soc., Perkin Trans. 1, 1991, 2702 Search PubMed.
- D. P. McAdam, A. M. A. Perera and R. V. Stick, Aust. J. Chem., 1987, 40, 1901 CAS.
- J. L. Gómez-Ariza, D. Sánchez-Rodas, I. Giráldez and E. Morales, Talanta, 2000, 51, 257 CrossRef CAS.
- S. N. Pedersen and K. A. Francesconi, Rapid Commun. Mass Spectrom., 2000, 14, 641 CrossRef CAS.
- W. R. Penrose, R. Black and M. J. Hayward, J. Fish. Res. Bd. Can., 1975, 32, 1275 Search PubMed.
- B. Bachmann, D. A. Hunter and K. A. Francesconi, Appl. Organomet. Chem., 1999, 13, 771 CrossRef CAS.
- A. D. Madsen, W. Goessler, S. N. Pedersen and K. A. Francesconi, J. Anal. At. Spectrom., 2000, 15, 657 RSC.
- V. W. M. Lai, W. R. Cullen and S. Ray, Mar. Chem., 1999, 66, 81 CrossRef CAS.
- J. S. Edmonds and K. A. Francesconi, J. Chem. Soc., Perkin Trans., 1983, 2375 Search PubMed.
- E. Larsen, Fresenius’ J. Anal. Chem., 1995, 352, 582 CrossRef CAS.
- Y. Shibata and M. Morita, Appl. Organomet. Chem., 1992, 6, 343 CrossRef CAS.
- E. H. Larsen, C. R. Quétel, R. Munoz, A. Fiala-Medioni and O. F. X. Donard, Mar. Chem., 1997, 57, 341 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |