Effects of electron withdrawing and donating groups on the efficiency of tris(2,2′-bipyridyl)ruthenium(II)/tri-n-propylamine electrochemiluminescence
David
Bruce
,
Jeff
McCall
and
Mark M.
Richter
*
Department of Chemistry, Southwest Missouri State University, Springfield, MO 65804, USA
First published on 11th December 2001
Abstract
The effects of electron withdrawing and electron donating groups on the electrochemiluminescent (ECL) properties of tris(2,2′-bipyridyl)ruthenium(II) (Ru(bpy)32+ where bpy = 2,2′-pyridine) are reported. The electrochemistry, photophysics and ECL of (bpy)2Ru(DC-bpy)2+, and (bpy)2Ru(DM-bpy)2+ (DC = 4,4′-dicarboxy-2,2′-bipyridine; DM = 4,4′-dimethyl-2,2′-bipyridine) have been studied relative to Ru(bpy)32+ in 50∶50 (v/v) acetonitrile(CH3CN)∶H2O (0.1 M KH2PO4), and aqueous solutions. Furthermore, the effects of Triton X-100 (polyethylene glycol tert-octylphenyl ether) on the electrochemical, spectroscopic and ECL properties of these compounds are reported. The anodic oxidation of Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+, and (bpy)2Ru(DM-bpy )2+ produces ECL in the presence of tri-n-propylamine (TPrA) in all solvent systems. ECL efficiencies (ϕecl, photons produced per redox event) of 0.73 and 0.84 for (bpy)2Ru(DC-bpy)2+, and (bpy)2Ru(DM-bpy)2+ were obtained in aqueous buffered solution, using Ru(bpy)32+ as a relative standard (ϕecl = 1.0). Addition of 0.4 mM Triton X-100 results in a greater than 2-fold increase in ECL efficiences (i.e., 3.8, 2.4 and 2.3 for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+, and (bpy)2Ru(DM-bpy)2+, respectively) using aqueous Ru(bpy)32+ containing no surfactant as standard (ϕecl = 1.0). ECL efficiencies of 27.4, 16.5 and 26.1 were found in 50∶50 (v/v) CH3CN∶H2O (0.1 M KH2PO4) for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+, and (bpy)2Ru(DM-bpy)2+, respectively, using aqueous Ru(bpy)32+ containing no surfactant as standard (ϕecl = 1.0). Detailed studies support adsorption of surfactant on the electrode surface, thus facilitating TPrA and ruthenium oxidation.
Introduction
Electrochemiluminescence (ECL) in aqueous micellar media is currently an area of active study.1–4 ECL involves the generation of excited states at an electrode and is a sensitive probe of electron and energy transfer processes at charged interfaces.5 Solubilization of Ru(bpy)32+ (bpy = 2,2-bipyridine) in aqueous solutions containing nonionic surfactants has led to significant, and potentially useful, changes in ECL properties. For example, increases in ECL efficiency (≥5-fold), oxidative current (≥2-fold) and duration of the ECL signal were observed in surfactant media upon oxidation of Ru(bpy)32+ and TPrA (TPrA = tri-n-propylamine).2,3,4 Oxidation of the coreactant TPrA is believed to form a strong reducing agent (e.g., TPrA˙) that can reduce Ru(bpy)33+ to *Ru(bpy)32+.6The mechanism of the surfactant effect is still unclear. However, recent work3,4 indicates that adsorption of Triton X-100 (polyethylene glycol tert-octylphenyl ether) and similar surfactants on Pt electrodes renders the surface more hydrophobic, facilitating coreactant oxidation and leading to increased ECL intensities in the Ru(bpy)32+–TPrA system. Electrode hydrophobicity on TPrA oxidation and the intensity of the Ru(bpy)32+–TPrA ECL system have also been examined.3 Various thiol molecules were used to modify Pt and Au electrode surfaces, with the hydrophobicity of the electrode controlled by the terminal group of the thiol monolayer (e.g., HO(CH2)6SH vs. CH3(CH2)5SH). The oxidation rate, and ECL intensity, were found to be much larger at modified electrodes with more hydrophobic surfaces.3
It is also possible to vary the hydrophobicity of the ECL luminophore (i.e., Ru(bpy)32+) by attaching a substituent to one, or more, of the bipyridyl rings (e.g., 4,4′-dimethyl-2,2′-bipyridine). Due to the analytical importance of Ru(bpy)32+ ECL in clinical analyses (e.g., immunoassays and DNA probes)7 and the improved detection limits and sensitivity of Ru(bpy)32+ in the presence of nonionic surfactants we decided to further investigate the nature of their interactions.
Therefore, the effects of electron withdrawing and electron donating groups on the electrochemiluminescent (ECL) properties of Ru(bpy)32+–TPrA are described in mixed solvent (acetonitrile–water) , aqueous and aqueous surfactant solutions.
Experimental
Materials
(bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ (DC = 4,4′-dicarboxy-2,2′-bipyridine; DM = 4,4′-dimethyl-2,2′-bipyridine) were prepared and characterized by literature methods.8 All other materials were used as received. Ru(bpy)3Cl2·6H2O (98%, Strem Chemical Inc, Newbury Port, MA), potassium phosphate monobasic hydrate (99%, EM Science, Gibbstown, NJ), tri-n-propylamine (TPA, 98%, Avocado Research Chemicals, Ward Hill, MA), and Triton X-100 (Avocado Research Chemicals). Potassium phosphate buffer solutions, 0.20 M KH2PO4·7H2O , were prepared with deionized water that had been passed through a Barnstead/Thermolyne filtration system. Buffer solutions containing TPrA (0.05 M) were prepared similarly except that it was necessary to stir them vigorously to completely dissolve the amine. The pH of these buffer solutions was adjusted with either 6 M HCl or 6 M H2SO4.Methods
Electrochemical, ECL, photoluminescence and UV-Vis instrumentation and experimental methods have been described elsewhere.2,4 All electrochemical and ECL experiments were referenced with respect to Ag/AgCl gel electrode (0.20 V vs. NHE).9 The working electrode was cleaned prior to each experiment by repeated cycling (+2.0 to −2.0 V) in 6.0 M sulfuric acid, followed by sonication in 2 M nitric acid and rinsing in deionized water.Solutions used to measure ECL intensities and ECL intensity versus surfactant concentrations included 10−6 to10−7M Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+, or (bpy)2Ru(DM-bpy)2+ and 0.05M TPrA in aqueous (0.2M KH2PO4 as electrolyte) or mixed 50∶50 (v/v) H2O∶CH3CN (0.1M KH2PO4 as electrolyte) solution. Surfactant solutions varied from 0.1 mM to 1.0 mM in Triton X-100. ECL was measured by sweeping from 0 to +2.0 V versus Ag/AgCl at 0.1 V s−1 using cyclic voltammetry.
For surfactant adsorption studies (i.e., Dip Tests) two solutions were used. Each contained 10−7 M ruthenium complex and 0.05 M TPrA. One of these solutions contained 0.4–0.5 mM Triton X-100 while the other contained no surfactant. The working electrode was immersed in one of these ‘Dip’ solutions for 10 min, rinsed gently with H2O for one minute to remove any unadsorbed species and placed in a solution of 0.1 μM ruthenium compound and 0.05 M TPrA in 0.2 M potassium phosphate buffer with no surfactant. ECL was measured by sweeping from 0 to +2.0 V versus Ag/AgCl at 0.1 V s−1 using cyclic voltammetry.
Photoluminescence and ECL spectra were obtained with a Shimadzu RF-5301 spectrofluorophotometer (slit widths 5 nm) on solutions containing 10−3–10−4 M ruthenium compound with 0.05 M TPA, in 0.2 M KH2PO4 or 50∶50 CH3CN∶H2O (0.1 M KH2PO4). Photoluminescence excitation was at 454 nm with detection between 500 and 700 nm. ECL efficiencies (ϕecl = photons generated per redox event) were obtained by the literature methods,10 using Ru(bpy)32+ (ϕecl = 1) as the standard. Similarly, relative photoluminescence efficiencies followed published procedures11 using Ru(bpy)32+ (ϕem (H2O) = 0.042).
Results and discussion
Electrochemistry
Cyclic voltammetric (CV) data for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ in the absence of surfactant are presented in Table 1. The oxidative wave is assigned to the Ru2+/3+ couple.11 As expected, incorporation of electron withdrawing and donating substituents shifts the Ru2+/3+ wave to higher and lower potentials, respectively, compared to Ru(bpy)32+. Peak potentials are also ∼100 mV more anodic in mixed solvent solutions showing the sensitivity of these systems to the solvent environment.12 Cyclic voltammograms are reversible to quasi-reversible in nature with ia/ic ≤1.39, and a peak-to-peak separation (ΔEpp) of ∼70mV. ΔEpp for the ruthenium compounds is less than or equal to that observed for ferrocene+/0 under similar conditions.| aSolvent | b E°/V | b E pp/V | i a/ic | |
|---|---|---|---|---|
| a H2O refers to aqueous solution with 0.2 M KH2PO4 as electrolyte. 50∶50 refers to mixed aqueous–non-aqueous solution of 50∶50 (v/v) CH3CN∶H2O (0.1 M KH2PO4 as electrolyte). b Volts vs. Ag/AgCl. E° refers to the standard potential. Epp refers to the difference between anodic and cathodic peak potentials in cyclic voltammetry. ia/ic correspond to the anodic and cathodic peak currents, respectively, in cyclic voltammetry. | ||||
| Ru(bpy)32+ | H2O | 1.039 | 0.061 | 1.14 |
| Ru(bpy)32+ | 50∶50 | 1.137 | 0.065 | 1.14 |
| (bpy)2Ru(DM-bpy)3+ | H2O | 0.9815 | 0.059 | 0.93 |
| (bpy)2Ru(DM-bpy)3+ | 50∶50 | 1.103 | 0.069 | 1.32 |
| (bpy)2Ru(DC-bpy)3+ | H2O | 1.077 | 0.065 | 1.39 |
| (bpy)2Ru(DC-bpy)3+ | 50∶50 | 1.150 | 0.075 | 1.23 |
The redox potentials of Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ are nearly independent of the presence and concentration of Triton X-100 surfactant in aqueous phosphate buffer solution. This confirms previous results for Ru(bpy)32+ in the presence of a series of Triton surfactant molecules.2,4 The lack of an oxidative potential shift implies that there is not a stronger interaction between Ru(bpy)33+, (bpy)2Ru(DC-bpy)3+ and (bpy)2Ru(DM-bpy)3+ and surfactant media compared to the reduced (i.e., 2+) forms of these complexes.13,14
Absorption and photoluminescence
Absorption spectra for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ in aqueous solution are shown in Fig. 1, and the data for both aqueous and 50∶50 (v/v) CH3CN∶H2O are presented in Table 2. The absorption bands centered around 450 nm have been assigned as MLCT (metal-to-ligand charge transfer) transitions.11,12 The visible absorption bands are not solvent dependent. Excitation into the broad visible absorption band (λmax ≅ 454 nm) produces room temperature photoluminescence characteristic of MLCT transitions for all three luminophores (Fig. 2 and Table 2). Emission bands are 596, 629 and 605 nm for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+, respectively, in H2O, and 598, 624 and 606 nm in 50∶50 (v/v) CH3CN∶H2O. The wavelengths and intensities of emission maxima in aqueous solution show no dependence on Triton X-100 concentration from 0.1–1 mM suggesting only a weak interaction between these species. This is not surprising since excited state lifetime studies13,14 indicate that the binding between Ru(bpy)32+ and Triton X-100 is van der Waal’s or hydrophobic in nature.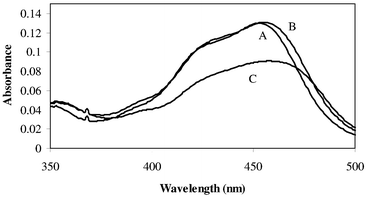 | ||
| Fig. 1 Absorption spectra of (A) Ru(bpy)32+, (B) (bpy)2Ru(DM-bpy)2+ and (C) (bpy)2Ru(DC-bpy)2+ containing 0.1 mM ruthenium complex in aqueous buffered (0.2M KH2PO4) solution at pH 8. | ||
| aSolvent | λ abs/nm | λ em/nm | b ϕ em | c ϕ ecl | c d ϕ ecl-Triton | |
|---|---|---|---|---|---|---|
| a H2O refers to aqueous solution with 0.2 M KH2PO4 as electrolyte. 50∶50 refers to mixed aqueous–non-aqueous solution of 50∶50 (v/v) CH3CN∶H2O (0.1 M KH2PO4 as electrolyte). b Relative photoluminescence emission efficiencies using Ru(bpy)32+(H2O) as standard. c Relative ECL efficiencies using Ru(bpy)32+ (H2O or 50∶50) as standard. Value is average of at least three points with a standard deviation of ± 0.05. d [Triton X-100] = 0.4 mM. Value is the average of at least three points with a standard deviation of ± 0.05. | ||||||
| Ru(bpy)32+ | H2O | 454 | 596 | 0.042 | 1.0 | 3.8 |
| Ru(bpy)32+ | 50∶50 | 453 | 598 | 0.031 | 1.0 | N/A |
| (bpy)2Ru(DM-bpy)2+ | H2O | 455 | 605 | 0.030 | 0.84 | 2.3 |
| (bpy)2Ru(DM-bpy)2+ | 50∶50 | 455 | 606 | 0.025 | 0.95 | N/A |
| (bpy)2Ru(DC-bpy)2+ | H2O | 458 | 629 | 0.020 | 0.73 | 2.4 |
| (bpy)2Ru(DC-bpy)2+ | 50∶50 | 458 | 624 | 0.024 | 0.60 | N/A |
 | ||
| Fig. 2 Photoluminescent emission spectra of (A) Ru(bpy)32+, (B) (bpy)2Ru(DM-bpy)2+ and (C) (bpy)2Ru(DC-bpy)2+ containing 0.1 mM ruthenium complex in aqueous buffered (0.2 M KH2PO4) solution at pH 8. | ||
Electrochemiluminescence
Due to the reversible nature of the Ru2+/3+ anodic redox couple TPrA was used as an ‘oxidative-reductive’ coreactant6 to generate ECL. ECL was observed for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ in 50∶50 (v/v) CH3CN∶H2O and aqueous solutions at a Pt interface. The ECL intensity peaks at potentials of ∼+1.2 V for CH3CN∶H2O and aqueous solutions (Fig. 3). At these potentials, oxidation of both TPrA (Ea ∼+0.5 V vs. Ag/AgCl) and Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ or (bpy)2Ru(DM-bpy)2+ has occurred indicating that the ECL emission is due to the MLCT excited state of the ruthenium compounds. ECL spectra were obtained for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ in each solvent and representative examples are shown in Fig. 4. The ECL spectra are nearly identical to photoluminescence spectra and indicate formation of the same MLCT excited states in both experiments. ECL efficiencies (ϕecl, photons generated per redox event) were calculated using Ru(bpy)32+ as a relative standard (taken as 1 in H2O (0.2 M KH2PO4))15 and correspond fairly well to the relative photoluminescence efficiencies (Table 2). Interestingly, ϕecl values in 50∶50 (v/v) CH3CN∶H2O are much higher than in aqueous solution (e.g., ϕecl of Ru(bpy)32+ (50∶50) = 27.4, (bpy)2Ru(DC-bpy)2+ (50∶50) = 16.5 while ϕecl of (bpy)2Ru(DC-bpy)2+ (50∶50) = 26.1 if the integrated ECL intensity is compared using Ru(bpy)32+ (H2O) as a relative standard (ϕecl = 1)). Using Ru(bpy)32+ (50∶50) as a standard with ϕecl = 1 results in ϕecl of (bpy)2Ru(DC-bpy)2+ (50∶50) = 0.84 while ϕecl of (bpy)2Ru(DC-bpy)2+ (50∶50) = 0.73. The differences among solvent systems may be due to interactions of the excited state (e.g., *Ru(bpy)32+), or the strong reducing agent (TPrA˙) with solvent molecules. | ||
| Fig. 3 ECL intensity versus potential for (A) Ru(bpy)32+, (B) (bpy)2Ru(DM-bpy)2+ and (C) (bpy)2Ru(DC-bpy)2+ containing 1μM ruthenium compound, and 0.05 M TPrA in aqueous solution (0.2 M KH2PO4) at pH 8. | ||
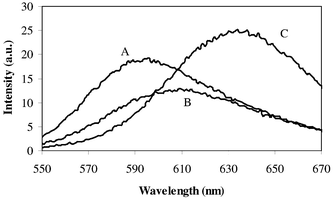 | ||
| Fig. 4 Electrochemiluminescence spectra of (A) Ru(bpy)32+, (B) (bpy)2Ru(DM-bpy)2+ and (C) (bpy)2Ru(DC-bpy)2+ containing 0.1 mM ruthenium complex in aqueous buffered (0.2 M KH2PO4) solution at pH 8. | ||
Fig. 5 illustrates the relationship between surfactant concentration and ECL intensity among the three ruthenium compounds. At concentrations well below the critical micelle concentration (cmc = 0.333 mM for Triton X-100) increased ECL intensity is observed. This indicates interactions between surfactant molecules, ECL luminophore and/or TPrA.1,2,4 The general trend observed in Fig. 5 is an increase in ECL signal until approximately 0.4 mM surfactant. At this point ECL signal changes at a much slower rate for all three compounds. Such dramatic increases cannot be attributed solely to solubilization of Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ or (bpy)2Ru(DM-bpy)2+ in micelles since previous studies in the presence of Triton surfactants with varying cmcs showed similar trends.4
 | ||
| Fig. 5 ECL intensity (counts per second, cps × 105) vs. concentration of surfactant for Ru(bpy)32+ (●), (bpy)2Ru(DM-bpy)2+ (■), (bpy)2Ru(DC-bpy)2+ (▲). 0.1 μM Ru(bpy)32+, 0.2 M KH2PO4, 0.05 M TPrA. Each point is the average of at least three scans with a standard deviation of ± 5%. | ||
Regardless of the mechanism of the surfactant effect, dramatic increases of greater than 2-fold in ECL intensity are observed for the ruthenium complexes in the presence of 0.4 mM Triton X-100 (Table 2). If, as previous studies have indicated,3,4 adsorption of Triton X-100 renders the electrode more hydrophobic, one might expect changes in the hydrophobicity of Ru(bpy)32+ to lead to dramatic increases and/or decreases in ECL intensity. Such changes have been observed for Ru(bpy)32+/TPrA ECL when various thiol monolayers were adsorbed on electrode sufaces.3 The oxidation rate of TPrA and Ru(bpy)32+, as well as the ECL intensity were found to be much larger at modified electrodes with more hydrophobic surfaces. ECL efficiencies for Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ in 0.3 mM surfactant solution are 3.8, 2.3 and 2.4 (Table 2), respectively, compared to Ru(bpy)32+ with no surfactant (ϕecl = 1). However, incorporation of both hydrophobic (i.e., methyl) and hydrophilic (i.e., carboxylate) derivates onto one of the bpy ligands in each complex does not lead to a dramatic increase for the dimethyl derivative, as one might expect due to its increased hydrophobicity compared to Ru(bpy)32+ and (bpy)2Ru(DC-bpy)2+. In fact, the magnitude of the increases in (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ correspond fairly well with ϕecl when no surfactant is present, suggesting that factors other than adsorption of the ruthenium complex into the surfactant layer, such as the adsorption and oxidation of TPrA, play a major role in the ECL.
To confirm the effect of surfactant adsorption at the electrode surface for all three complexes, a platinum electrode was immersed in 0.5 mM surfactant for 10 min, rinsed with water, and then placed in a solution containing TPrA and ruthenium compound. Cyclic voltammetric results are presented in Table 3, and ECL results in Fig. 6. In the first cycle, ECL intensities and oxidation currents were larger for surfactant containing solutions. During subsequent cycles, however, both current and ECL intensities dropped dramatically. By the 3rd cycle ECL intensities and currents of the surfactant dipped electrodes were equivalent to those observed at bare electrodes. Since the increased surface charge on the electrode at higher potentials will lead to desorption of the surfactants, this clearly indicates that the adsorption of surfactant species at the electrode/solution interface plays a role on the ECL of Ru(bpy)32+, (bpy)2Ru(DC-bpy)2+ and (bpy)2Ru(DM-bpy)2+ and TPrA in surfactant solution. The increase of oxidation current compared to a bare electrode also indicates the formation of a surfactant adsorption layer on the Pt surface. This layer may allow for more luminophore and TPrA to be oxidized giving rise to the higher ECL intensities.
| Compound | Surfactant | aSweep 1 | aSweep 2 | aSweep 3 | ||||
|---|---|---|---|---|---|---|---|---|
| I ECL/cps × 104 | i a/mA | I ECL/cps × 104 | i a/mA | I ECL/cps × 104 | i a/mA | |||
| a Anodic oxidation current (ia) measured at the potential where the ECL peak appeared. Standard deviation of ECL measurements is ±5%; cps = counts per second.A full cyclic voltammetric sweep (e.g., Sweep 1) was from 0.0 V to +2.0 V to +0.0 V vs. Ag/AgCl. | ||||||||
| Ru(bpy)32+ | None | 2.24 | −4.89 | 0.380 | −2.96 | 0.316 | −1.78 | |
| Ru(bpy)32+ | 0.5 mM | 5.62 | −10.9 | 0.844 | −7.44 | 0.389 | −5.94 | |
| (bpy)2Ru(DM-bpy)2+ | None | 1.60 | −4.11 | 0.282 | −1.79 | 0.254 | −1.21 | |
| (bpy)2Ru(DM-bpy)2+ | 0.5 mM | 4.63 | −6.58 | 0.283 | −2.60 | 0.251 | −1.60 | |
| (bpy)2Ru(DC-bpy)2+ | None | 1.96 | −4.30 | 0.300 | −1.78 | 0.249 | −1.40 | |
| (bpy)2Ru(DC-bpy)2+ | 0.5 mM | 3.38 | −6.91 | 0.326 | −4.25 | 0.282 | −3.1 | |
 | ||
| Fig. 6 ECL intensity versus identity of ruthenium compound for adsorption study (i.e. ‘Dip Test’). Platinum electrode immersed in 0.4 mM surfactant solution for 10 min, rinsed, and placed in a separate solution containing 0.1 μM ruthenium compound, 0.2 M KH2PO4, 0.05 M TPrA at pH 8. Each point is the average of at least three scans with a standard deviation of ± 5%. | ||
Conclusions
This work clearly shows that the surfactant effect on the ECL of ruthenium complexes is not limited to Ru(bpy)32+. It also clearly shows that adsorption of surfactant plays a role in the ECL process since experiments show that an adsorption layer forms on the electrode surface allowing for increased ruthenium and/or TPrA oxidation. Surprisingly, the hydrophobicity of the ECL luminophore does not lead to dramatic increases or decreases in ECL efficiency, suggesting that adsorption of TPrA or other factors contribute to the surfactant effect. Studies on other ruthenium derivatives (e.g., (DC-bpy)2Ru(bpy)2+ and (DC-bpy)3Ru2+) in the presence and absence of surfactants will be the subject of a forthcoming report.Acknowledgement
Acknowledgement is made to the donors of the American Chemical Society-Petroleum Research Fund for partial support of this research. We also gratefully acknowledge Southwest Missouri State University for its financial support.References
- (a) P. McCord and A. J. Bard, J. Electroanal. Chem., 1991, 318, 91 CrossRef CAS; (b) J. Ouyang and A. J. Bard, Bull Chem. Soc. Jpn., 1988, 61, 17 CAS; (c) S. K. Lee and A. J. Bard, Anal. Lett., 1998, 31, 2209 CAS.
- S. Workman and M. M. Richter, Anal. Chem., 2000, 72, 5556 CrossRef CAS.
- Y. Zu and A. J. Bard, Anal. Chem., 2001, 73, 3960 CrossRef CAS.
- B. Factor, B. Muegge, S. Workman, E. Bolton and J. Bos, Anal. Chem, 2001, 73, 4621 CrossRef CAS.
- (a) L. R. Faulkner and A. J. Bard, in Electroanaytical Chemistry, ed A. J. Bard, Marcel Dekker, New York, 1977, vol. 10, pp. 1–95; Search PubMed; (b) L. R. Faulkner and R. S. Glass, in Chemical and Biological Generation of Excited States, ed. W. Adam and G. Cilento, Academic Press, New York, 1982, p. 191 Search PubMed.
- J. K. Leland and M. J. Powell, J. Electroanal. Chem., 1991, 318, 91 CrossRef CAS.
- G. F. Blackburn, H. P. Shah, J. H. Kenten, J. Leland, R. A. Kamin, J. Link, J. Peterman, M. J. Powell, A. Shah, D. B. Tulley, S. K. Tyagi, E. Wilkins, T.-G. Wu and R. J. Massey, Clin. Chem., 1991, 37, 1534 Search PubMed.
- (a) C. H. Brauenstein, A. D. Baker, T. C. Strekas and H. D. Gafney, Inorg. Chem., 1984, 23, 857 CrossRef; (b) D. P. Rillema and K. B. Mack, Inorg. Chem., 1982, 21, 3849 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- M. M. Richter, A. J. Bard, W. K. Kim and R. H. Schmehl, Anal. Chem., 1998, 70, 310 CrossRef CAS.
- (a) J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583 CrossRef CAS; (b) J. Van Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853 CrossRef CAS.
- (a) C. H. Braunstein, A. P. Baker, T. C. Strekas and H. D. Gafney, Inorg. Chem., 1984, 23, 857 CrossRef; (b) D. P. Rillema and K. B. Mack, Inorg. Chem., 1982, 21, 3849 CrossRef CAS; (c) E. M. Kober, J. P. Caspar, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4587 and references therein Search PubMed.
- K. Mandal, B. L. Hauenstein Jr., J. N. Demas and B. A. DeGraff, J. Phys. Chem., 1983, 87, 328 CrossRef CAS.
- W. J. Dressick, B. L. Hauenstein Jr., T. B. Gilbert, J. N. Demas and B. A. DeGraff, J. Phys. Chem., 1984, 88, 3337 CrossRef CAS.
- M. M. Richter, J. D. Debad, D. R. Striplin, G. A. Crosby and A. J. Bard, Anal. Chem., 1996, 68, 4370 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |