Methods for testing antioxidant activity
Michael
Antolovich
,
Paul D.
Prenzler
,
Emilios
Patsalides
,
Suzanne
McDonald
and
Kevin
Robards
*
School of Science and Technology, Charles Sturt University, Locked Bag 588, Wagga Wagga 2678, Australia. E-mail: krobards@csu.edu.au; Fax: 02 6933 2737; Tel: 02 6933 2547
First published on 23rd November 2001
 Kevin Robards Kevin Robards | Kevin Robards is Associate Professor of Chemistry at Charles Sturt University Riverina. He obtained his PhD in analytical chemistry from the University of New South Wales in 1979. His research interests are focused on the application of analytical chemistry to food science and in particular the identification and role of naturally occurring phenolic compounds in fruits. |
1 Introduction
The importance of oxidation in the body and in foodstuffs has been widely recognized. Oxidative metabolism is essential for the survival of cells. A side effect of this dependence is the production of free radicals and other reactive oxygen species that cause oxidative changes. There is increasing evidence for the involvement of such species in a variety of normal in vivo regulatory systems.1 When an excess of free radicals is formed, they can overwhelm protective enzymes such as superoxide dismutase, catalase and peroxidase and cause destructive and lethal cellular effects (e.g., apoptosis) by oxidizing membrane lipids, cellular proteins, DNA and enzymes, thus shutting down cellular respiration. Furthermore, reactive oxygen species seem to influence cell signalling pathways in ways that are only now being unravelled.2,3 Oxidation can also affect foods, where it is one of the major causes of chemical spoilage,4 resulting in rancidity and/or deterioration of the nutritional quality, colour, flavour, texture and safety of foods.5 It is estimated that half of the world’s fruit and vegetable crops are lost6 due to postharvest deteriorative reactions. Defence mechanisms against the effects of excessive oxidations are provided by the action of various antioxidants and the need to measure antioxidant activity is well documented.Methods of assessing antioxidant behaviour fall into two broad categories reflecting the focus on activity in foods or bioactivity in humans. In the case of food systems, the need is to assess the efficacy of an antioxidant(s) in providing protection for the food7 against oxidative spoilage. A sub-category involves measurement of activity in foods, particularly fruits, vegetables and beverages, but with a view to predicting dietary burden and in vivo activity.8,9 Oxidative stress in humans arises from an imbalance in the antioxidant status (reactive oxygen species versus defence and repair mechanisms). Among the endogenous defences are enzymes such as superoxide dismutase, catalase and glutathione peroxidase, plus vitamin E, uric acid and serum albumins. Besides these defences, consumption of dietary antioxidants is also important. An important distinction from food-based systems is the absence of a single, definable substrate in many instances in vivo.
This review examines the various methods of measuring antioxidant activity particularly as they relate to lipid oxidation. This should be distinguished from the related process of measuring the concentration of an antioxidant(s) which is not considered here. However, it should be recognized that the two are related as antioxidants generally exhibit pro-oxidant effects at higher concentration. The term ‘activity’ as applied to antioxidants needs clarification as it can have a variety of meanings. Relevant aspects include: mechanistic intervention, e.g., free radical scavenger, catalytic decomposition, pro-oxidant suppression; rate of scavenging, e.g., near-diffusion or controlled; medium or substrate selectivity (e.g., aqueous, surface or lipid phase); concentration effectiveness (moles of free radicals scavenged per mole of antioxidant); synergistic effect for other antioxidants.
However, the term seems to be loosely applied to identifying ‘activity’ as that measured by one or several common or standard tests such as listed in Table 1. In many cases neither a specific substrate nor a specific additive may be involved but extracts may be screened to identify those which exhibit antioxidant activity according to the test method(s) employed. For example, TLC screening may be used10,11 to identify components in extracts that exhibit such activity. It is also possible to use screening methods to identify the class of antioxidant (e.g., phenolic) or even its action12 by the use of spray reagents (e.g., complexing agent, radical inhibitor, hydroperoxide decomposer). In any case, such ‘activities’ need to be supported by testing in the actual substrate and conditions of interest. This is particularly important for in vivo testing where absorption, metabolic transformations, excretion,13 the presence of competitive enzymes and antioxidants in addition to pro-oxidants may profoundly affect the in vivo activity of test antioxidants.
| Test | Measurement | Units |
|---|---|---|
| Peroxide value | Peroxides and hydroperoxides | mequiv. kg−1of active oxygen |
| Diene conjugation | 1,4-Dienes produced by early stages in lipid autoxidation | Absorbance/unit mass mg kg−1 linoleic acid equivalents |
| Thiobarbituric acid reactive substances (TBARS) | Thiobarbituric acid derivatives of malondialdehyde absorbing at 532–535 nm | mg kg−1 (ppm w/w) as malondialdehyde |
| Kreis test | Phloroglucinol derivatives of malondialdehyde and other aldehydes absorbing at 546 nm | Red colour on Lovibond scale (empirical); mg kg−1 (ppm w/w) as malondialdehyde |
| Anisidine value | Aldehydes (mainly alkenals) | 100 times the corrected absorbance in a l cm cell at 350 nm containing l g of oil or fat per 100 mL of isooctane–acetic acid solvent |
| Hexanal formation, pentane formation, hexane formation, etc. | Specific oxidation end-product formed | mg kg−1 of product formed |
| ABTS•+ assay | Absorbance of radical cation in aqueous medium at 734 nm or other suitable wavelength | Inhibition time for appearance of radical cation under specified conditions or decay rate once formed |
| Total radical trapping antioxidant parameter (TRAP) | ||
| Phycoerythrin assay | Fluorescence intensity | Inhibition of fluorescence decay under specified conditions of autoxidation. Can be expressed as trolox equivalents? |
| Electron spin resonance (ESR) spin-trap test | Intensity/rates of change in concentration of antioxidant or spin-trap derivative radicals | mg L−1 of radical species (cf. stable standard such as di-tert-butyl nitroxide) |
| TG/DTA | Time required for development of autoxidation in a dynamic oxygen atmosphere at specified temperature | ΔT (°C), mass change (mg) |
In the case of natural antioxidants, they may be multifunctional. The mechanism that is operative or dominant in a particular situation is dependent on conditions and yet this will affect the kinetics and hence the antioxidant activity. These differences and particularly the variation in analytical procedures account for the inconsistent results that have been reported for a number of recognized antioxidants.14
An important distinction can be made between short- and long-term antioxidant protection. This is related to the reaction kinetics15,16 and the rate at which an antioxidant reacts with a specific radical versus the thermodynamics of the reaction and how completely the antioxidant reacts. For instance, disappearance of the DPPH radical (Table 2) followed a double-exponential equation in the presence of edible oils and oil fractions17 which suggested the presence of a fast- and slow-acting group of antioxidants.
| Acronymn | Name |
|---|---|
| AAPH | 2,2′-Azobis(2-amidinopropane)hydrochloride |
| ABTS | 2,2′-Azinobis(3-ethylbenzthiazoline)-6-sulfonic acid |
| AMVN | 2,2′-Azobis(2,4-dimethylvaleronitrile) |
| BHA | Butylated hydroxyanisole |
| BHT | Butylated hydroxytoluene |
| BNB | tert-Butylnitrosobenzene |
| CL | Chemiluminescence |
| DBNBS | 3,5-Dibromo-4-nitrosobenzenesulfonic acid |
| DMPO | 5,5-Dimethylpyrroline-N-oxide |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| FRAP | Ferric reducing antioxidant power |
| GC(-MS) | Gas chromatography(–mass spectrometry) |
| HPLC | High-performance liquid chromatography |
| LDL | Low-density lipoprotein |
| MDA | Malondialdehyde |
| ORAC | Oxygen radical absorbance capacity |
| POBN | α-(4-Pyridyl-1-oxide) N-tert-butylnitrone |
| PV | Peroxide value |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| TAA | Total antioxidant activity |
| TBA | Thiobarbituric acid |
| TBARS | Thiobarbituric acid reactive substances |
| TBHQ | tert-Butylhydroquinone |
| TEAC | Trolox equivalent antioxidant activity |
| TG/DTA | Thermogravimetry/differential thermal analysis |
| TRAP | Total radical trapping parameter |
Following a brief introduction to oxidative processes and the mechanism of antioxidant action, an historical background to activity tests is provided. The relationship of tests designed for food systems and their extension to physiological systems is presented. These may involve in vitro or in vivo testing and in the latter case may involve either invasive or non-invasive techniques. In vitro methods provide a useful indication of antioxidant activities but data obtained by these methods are difficult to apply to biological systems. On the other hand, in vivo measurements are difficult owing to problems relating to cellular uptakes of the antioxidants and the transport processes. Non-invasive techniques such as nuclear magnetic resonance (NMR) spectrometry may be useful but require relatively high antioxidant concentrations. The extensive literature concerning antioxidants precludes exhaustive treatment and rather selected examples of different tests have been chosen to illustrate various points. The present review complements that by Frankel and Meyer,18 which emphasizes the need for multi-faceted testing of antioxidant activity. For convenience, all acronyms used in this review are collected in Table 2.
2 Processes of lipid oxidation
A number of chemical and physical phenomena can initiate oxidation which proceeds continuously in the presence of a suitable substrate(s) until a blocking defence mechanism occurs. Target substances include oxygen, polyunsaturated fatty acids, phospholipids, cholesterol and DNA.19 Lipid oxidation is important in food deterioration and oxidative modification of low-density lipoprotein (LDL) (Table 2). Lipid oxidation proceeds20via three different pathways: (1) non-enzymatic free radical-mediated chain reaction, (2) non-enzymatic, non-radical photo-oxidation and (3) enzymatic reaction. An example of route (2) is the stoichiometric oxidation of oleic acid by singlet oxygen21,22 to produce two allylic hydroperoxides via addition of oxygen at either end of the double bond. The singlet oxygen is produced by sensitizers such as myoglobin or chlorophyll. Pathway (3) involves the action of lipoxygenases on various substrates.Pathway 1 is the classical free radical route23 that leads to initiation of rapidly progressing, destructive chain reactions. The essential features of oxidation via a free radical-mediated chain reaction are initiation, propagation, branching and termination steps.24 The process may be initiated by the action of external agents such as heat, light or ionizing radiation or by chemical initiation involving metal ions or metalloproteins.25
| Initiation: LH + R• → L• + RH |
| Propagation: L• + O2 → LOO• |
| LOO• + LH → L• + LOOH |
| Branching: LOOH → LO• + HO• |
| 2LOOH → LOO• + LO• + H2O |
| LOOH + Mn+ + H+ → LO• + M(n + 1)+ + H2O |
| LOOH + M(n + 1)+ + OH− → LOO• + Mn+ + H2O |
| Termination: LO• + LO• → |
| LOO• + LOO• → non-radical products |
| LO• + LOO• → |
There are obvious differences between the reactions occurring in vivo and in foods27–30 that may be exposed to elevated temperatures during storage and/or processing. For instance, hydroperoxides decompose readily and spontaneously at 160 °C and the peroxy radical concentration can become relatively high under such conditions, thus leading to the formation of polymers. Similarly, the reaction mechanism is different for emulsified and bulk lipids.27 The range of effects of free radicals is only a few ångströms, whereas the action of the non-free radical hydrogen peroxide is several nanometres and hydrogen peroxide can pass biological membranes freely. Nevertheless, there are essential features of the process that are similar in all cases. The measurement of antioxidant activity of certain components in vivo requires the definition of the type of free radical formation. At least four different types may be identified as: free iron and the Fenton reaction;31 mitochondrial lesions and pore reactions leading to apoptosis;32 chemically induced free radical formation (e.g. with paraquat);33 and hydrogen peroxide formation in vivo.34
3 Antioxidants
An antioxidant may be defined35 as ‘any substance that when present at low concentrations, compared with those of the oxidizable substrate, significantly delays or inhibits oxidation of that substrate’. For convenience, antioxidants have been traditionally divided into two classes, primary or chain-breaking antioxidants and secondary or preventative antioxidants.36 Secondary or preventative antioxidants are compounds that retard the rate of oxidation. This may be achieved in a number of ways including removal of substrate or singlet oxygen quenching.18 Primary antioxidants, AH, when present in trace amounts, may either delay or inhibit the initiation step by reacting with a lipid radical or inhibit the propagation step by reacting with peroxyl or alkoxyl radicals:36| L• + AH → LH + A• |
| LOO• + AH → LOOH + A• |
| LO• + AH → LOH + A• |
| A• + LOO• → LOOA |
| A• + LO• → LOA |
The activation energy of the above reactions5 increases with increasing A–H and L–H bond dissociation energy. Therefore, the efficiency of the antioxidant increases with decreasing A–H bond strength.
Chain-breaking antioxidants may occur naturally or they may be produced synthetically as in the case of BHT, BHA, TBHQ and the gallates. The synthetic antioxidants are widely used in the food industry20 and are included in the human diet.37,38 The use of naturally occurring antioxidants39 has been promoted because of concerns regarding the safety of synthetic antioxidants,40,41 with natural alternatives (e.g., plant biophenols) possessing antioxidant activity similar to or even higher than that of synthetic antioxidants.8,42
4 Measurement of antioxidant activity
Antioxidant activity cannot be measured directly but rather by the effects of the antioxidant in controlling the extent of oxidation. Methods show extreme diversity. Some methods involve a distinct oxidation step followed by measurement of the outcome as, for example, oxidation of linoleic acid followed by determination of diene conjugation. In other instances, there is no clear distinction between the various steps in the procedure.The features of an oxidation are a substrate,43 an oxidant and an initiator, intermediates and final products and measurement of any one of these can be used to assess antioxidant activity. For instance, in monitoring antioxidant activity in a food, potential measurements include PV,44 thiobarbituric acid value, iodine value, free fatty acid content, polymer content, viscosity, absorption at 232 and 268 nm, colour, fatty acid composition and ratio of unsaturated to saturated fatty acids (e.g., C18:2/C16:0). Physiological activity can be assessed by in vitro measurements such as the susceptibility of isolated LDL to oxidation.45,46 However, the preferable approach involves in vivo measurement of LDL oxidation products such as hydroxy-fatty acids or oxysterols or indirect indicators of lipid oxidation (e.g., F-2-isoprostanes).47–49 Alternatively, the immunological response to antigenic lipid oxidation products can be measured.
In studying antioxidant activity, the source of ROS and the target substrate must always be considered. An antioxidant may protect lipids against oxidative damage whilst accelerating damage to other biological molecules.50 Thus, Aruoma et al.50 used several measures of antioxidant activity and posed a series of questions that serve as a guide in evaluating antioxidant efficacy. The use of a number of different measures of activity is becoming a feature of published studies.18,51
Most test procedures use accelerated oxidation involving an initiator to manipulate one or more variables in the test system. Initiators include increased temperature and partial pressure of oxygen, addition of transition metal catalysts,52 exposure to light to promote photosensitized oxidation by singlet oxygen,53 variable shaking to enhance reactant contact16 and free radical sources.54 However, oxidation mechanisms can change as temperatures are raised55 while substrate effects56 and analytical technique57,58 also influence the results. The activity of an antioxidant on β-carotene will not be the same as on vegetable oil.59 The effect of substrate can be attributed to the strong influence of the unsaturation type and degree of the lipid system60 on the kinetics and mechanism of the antioxidative action. Photosensitized acceleration underestimates the effects of chain-breaking antioxidants.18
Metal ions such as copper and iron are the most common initiators in both food and biological systems. These ions catalyse the initiation and decomposition of hydroperoxides61 resulting in high levels of volatile decomposition products. Antioxidant effectiveness in an in vitro LDL oxidation test62 varied greatly with the level of copper ions used as catalyst.
The use of a substrate is considered essential18 and tests such as the ABTS assay that generally do not include a substrate are artificial and do not adequately mimic the processes in food and biological systems. After the substrate is oxidized under standard conditions, either the extent or rate of oxidation (an end-point) is measured by chemical, instrumental or sensory methods. Hence the essential features of any test are an oxidation initiator, a suitable substrate and an appropriate measure of the end-point. In rare instances, an initiator has been omitted and the scavenging of endogenous pre-formed hydroperoxides has been studied.63 The combinations of initiation, substrate and end-point that have been used are numerous and even with the same reagents, several analytical strategies are possible.64 These include (1) measurement at a fixed time point, (2) measurement of reaction rate, (3) lag phase measurement and (4) integrated rate measurement. In systems 1 and 2, the reagents are mixed and the end-point is measured after a pre-determined time interval in 1, whereas in 2, the rate of the reaction is monitored. In both cases, the presence of antioxidant in the reaction mixture reduces the change in end-point parameter. In system 3, the length of the lag time to end-point change is measured; samples with higher antioxidant activity suppress the change far longer than those with less activity. System 4 involves integration of the end-point versus time curve and is used where the reaction kinetics are not of a simple order.
Lipid substrates have included various oils and fats,65 linoleic acid,66 fatty acid methyl esters67 and LDL.68 In the case of oils/fats, the more bland materials are usually employed69 and these preferably only after refining and deodorizing. γ-Tocopherol at a concentration of 11 μg g−1 decreased70 hydroperoxide and secondary product formation to 46 and 39%, respectively. This has important implications as the potential for synergism with residual materials in a refined oil always exists and has led to the use of model substrates. Various model substrates have been described including methyl linoleate,71 linoleic acid66 and methyl linoleate in silicone oil.71 Citronellal was recently used72,73 as a substrate in an accelerated test based on measurement of its degradation product by gas chromatography. Nevertheless, model substrates are not without problems, not the least of which is duplicating actual conditions of use. LDL represents an obvious substrate and many in vitro tests have been described68,74,75 that exploit various end-points including measurement of conjugated dienes and hexanal. Despite extensive use, LDL is a very dubious substrate, since the vitamin E level in LDL may be an important factor for protection of peroxidation of the unsaturated fatty acid in LDL. Caution is necessary when extrapolating from in vitro tests on food components, or especially ill-defined extracts, to the human in vivo situation as antioxidant activity is a complex interplay of several related factors. Moreover, there is a distinction between antioxidant activity and the antioxidant capacity (i.e., the sum of all antioxidant activities of a mixture containing many antioxidants, e.g., serum) that this confers on the blood plasma and the effect on oxidative susceptibility, for example, of LDL. In this context, the morphology of the LDL particle is important and differences in antioxidant activity can often be rationalized in terms of partition coefficients and accessibility to the lipid peroxyl radicals.76 A considerable amount of evidence is accumulating to suggest that synergism between aqueous and lipophilic systems is the important factor77 (and this shift in attitude is reflected in a wholistic approach to the Mediterranean diet.78 For this reason, where the interest is in the relative bioactivity of an antioxidant, tests should be performed in both aqueous and lipophilic phase systems.63 Antioxidant activity in the lipophilic phase is a composite response to partitioning behaviour and rates of reaction with the relevant radical species. The kinetics of the various reactions need to be considered as most radicals are highly reactive species and can diffuse only very short distances.79 Data on the lipophilic phase derive from studies on fatty acids, liposomes,80,81 which have been used extensively as in vitro cellular models for investigating antioxidant activity and especially LDL. Several studies have examined structure–activity relationships82–87 and Rice-Evans et al.88 have presented a detailed discussion of structure–activity effects in both lipophilic and aqueous phases, the latter based on measurement of TEAC.
There is a need to exercise caution in the interpretation of data and to measure a number of oxidation parameters18 to evaluate antioxidant activity better. The activity of carnosine, a dipeptide, which is a useful antioxidant in food systems, has been carefully examined with large differences in the results in model systems.89 On the basis of MDA release in a liposome system, carnosine exhibited good antioxidant activity during methylene blue photosensitized oxidation, weak antioxidant activity during riboflavin 5′-phosphate sensitized oxidation and even a pro-oxidant effect during copper(II)-catalysed oxidation. The antioxidant effect in liposomes decreased89 according to the catalyst in the following order: copper/ascorbate, iron/ascorbate, hydrogen peroxide activated haemoglobin, photoactivated riboflavin and lipoxygenase. In the case of rosemary extracts, antioxidant effectiveness was significantly influenced by the type of system tested (bulk oils versus oil-in-water emulsions), by the oil substrates, the methods used to follow oxidation and the concentrations of test compounds.90 Ethanol has exhibited antioxidant activity in certain circumstances68 and this must be considered when measuring the antioxidant activity of alcoholic beverages or when lipophilic compounds have to be added as ethanolic solutions to a test substrate.91
Results are expressed in a variety of ways that make comparisons difficult.
4.1 Expression of results
Methods of expressing antioxidant activity appear to be as varied as the methods of measurement.92 All, however, attempt to indicate the effectiveness of substances to hinder the oxidation of a substrate under specified conditions. A practical measure of activity must show at least two things: whether the test substance has a detectable antioxidant or pro-oxidant effect under the test conditions; and a comparison of the quantitative effect or likely effect, of specified concentrations of different test materials on the substrate.Most methods for reporting activities are based on measurements using common test procedures such as those summarized in Table 1. These, in turn, involve direct or indirect measurement of the rate or extent of: (a) decay of substrate or probe substance or of oxygen consumption; (b) formation of oxidation products; or (c) formation or decay of probe free radicals.
In (a) and (b) antioxidant activity, whatever the mechanism, is demonstrated as an inhibitory effect on the extent or rate of consumption of reactants or the formation of products. Qualitative measures used in screening tests would be reported as ‘shows antioxidant activity’, ‘shows pro-oxidant activity’ or ‘shows no activity’ according to the test procedure. For quantitive measures most authors report activities as comparative results, e.g., peroxide values, TBARS assays or absorbance increase at 230–235 nm after a fixed time period, e.g., induction times. However, there appear to be no standard units for reporting such activity (efficiency, effectiveness, assay, capacity, action, etc.) independent of the test procedure. Antioxidant activity (AA) is, of course, a function of many parameters:
| AA |
| = |
| f |
| (time or rate; temperature; substrate; concentration of antioxidant; concentration of other substances, |
| e.g. |
| , oxygen, |
| peroxides |
| or other antioxidants/pro-oxidants, |
| etc |
| .; partitioning behaviour) |
| AA = (t - tREF)/[AH]tREF |
| RAA1 = AA1/AAREF |
| AA1 = RAA1 × AAREF |
The advantage of this definition is that common test methods such as those listed in Table 1 can be used to calculate activities in standard concentration terms based on the general methods described in Table 3.
| Method | Results |
|---|---|
| Induction time | h, d |
| Time to reach a set level of oxidation (pre-induction period) | h, d |
| Rate of oxidation (pre-induction period) | mol kg−1 hr−1, g L−1 d−1 |
| Concentration to produce equivalent effect to reference antioxidant (pre-induction period) | mol kg−1, g L−1 |
| Concentration of functional group after set time period | mequiv. kg−1 |
| Concentration of oxidation product after set time period | mg kg−1 (ppm w/w) |
| Scale reading after set time period | Absorbance, conductivity, etc. |
The third method of measuring antioxidant activity (c) assumes that oxidation is inhibited largely by the capture of initiating or propagating free radicals in autoxidation. They therefore focus on monitoring the capacity of additives/extracts for radical capture or inhibition of radical formation rather than on monitoring the actual oxidation itself. They form the basis of the newer test methods such as the ABTS/TEAC, DPPH radical and phycoerythrin assays. A variety of new parameters for expressing results therefore are used (see Table 4) which more or less serve the same purpose as those based on monitoring the extent of autoxidation. A high correlation should therefore exist between results for the two broad methods though this has still to be clearly demonstrated.
| Method | Results |
|---|---|
| Free stable radical quenching | Percentage inhibition |
| Free stable radical quenching | EC50, concentration to decrease concentration of test free radical by 50% |
| Free stable radical quenching | T EC50,time to decrease concentration of test free radical by 50% |
| Total radical-trapping antioxidant parameter (TRAP) | μmol peroxy radical deactivated L−1 |
| ABTS assay, phycoerythrin assay | TEAC (mM Trolox equivalent to 1 mM test substance) |
| Phycoerythrin assay | ORAC, oxygen radical absorbance capacity; μmol of Trolox equivalents |
| FRAP assay | Absorbance of Fe(II) complex at 593 nm produced by antioxidant reduction of corresponding tripyridyltriazine Fe(III) complex |
5 Individual procedures
Various chemical and physico-chemical procedures are used to monitor oxidation processes. One approach is to examine directly free radical production and its inhibition by antioxidants. In the more usual approach, various indirect measurements are used to assess the effectiveness of an antioxidant in preventing oxidative damage. These are based on measurement of the inhibition of the various intermediates or final reaction products of oxidation. Individual measurement of the antioxidant activity of all components in a sample is possible, but this can be time consuming and expensive. In addition, there may be synergism between antioxidants and examining one in isolation may not accurately reflect their combined action.93 It is therefore of interest to measure the TAA,94 which can be quantified by defining the amount of a suitable standard needed to produce the same end-point as the compound or material being analysed.95The desirable features of a test of antioxidant activity are the use of a substrate and conditions in the test that mimic the real situation and the ability to quantify the result by reference to a suitable standard. For instance, it follows from the definition of an antioxidant that its test concentration must be significantly lower than that of the substrate.
The chemistry of each of the more common procedures is described, with a brief historical overview of the development of the method and its applications to food and/or biological systems as appropriate. Finally, any problems associated with the procedure are highlighted.
5.1 Accelerated stability tests
Stability tests on edible oils and fats such as the Rancimat,55 Active Oxygen Method and Schaal oven test commonly involve accelerated deterioration tests,96,97 sometimes as a result of the action of light or UV radiation, but much more commonly at elevated temperatures. Heating an oil and periodically testing for weight gain was one of the oldest methods for evaluating oxidative stability.98 This can be used as a general method for antioxidant activity by selecting a pure substrate (e.g., tripalmitin or triolein) or other substrate and adding an antioxidant.99 This requires simple equipment and indicates directly oxygen consumption although the mass change may reflect other volatiles. The latter can be removed from the sample by pre-heating in an inert atmosphere. The technique can be extended to more sophisticated continuous monitoring of mass and energy changes as in thermogravimetry/differential scanning calorimetry.These accelerated tests are specific to the analysis of oxidation in foods with results usually expressed as an induction time. Such tests are often highly relevant to the conditions to which oils and fats are subject, as in production processing, food manufacture or domestic use.55 The usual substrates include lard, edible oils100 or a model substrate such as methyl linoleate.101 Following oxidation, the end-point is determined by measuring parameters such as conductivity, peroxide value or diene conjugation. The addition of an antioxidant results in the inhibition of oxidation. Results are quantified by measuring the induction time of a control and sample, with longer induction indicating better antioxidant activity.101,102
Antioxidant activity of grape extract in refined soybean oil was determined69 by the Rancimat and Schaal oven test in conjunction with PV determination. Results from the two accelerated tests were similar. There was also a good correlation (r = 0.9702, P < 0.05) between the antioxidant activity of an Apsergillus extract101 measured by Rancimat and a linoleic acid oxidation system using the thiocyanate method. This is frequently not the case and the relative activity of several synthetic and natural antioxidants differed when determined by Rancimat or a procedure entailing milder test conditions (lower temperature, no active aeration)103–105 or sunflower oil thin films in an accelerated oven test.104 Similarly, the trends in antioxidant activity differed106 according to whether hydroperoxide formation (PV) or decomposition (hexanal and volatiles) was measured in accelerated stability tests on olive oil. These differences are not uncommon,97 particularly with extracts of low to intermediate antioxidant activity. Stability tests and their limitations have been reviewed by Frankel,107 who summarized some of the published literature on the methods used in the evaluation of various natural antioxidants.
There is intense interest in identifying natural antioxidants for use in foods and there has been considerable focus39,103,108,109 on plant biophenols. It was estimated110 from Rancimat data that o-diphenols contributed over 50% to the stability of virgin olive oil. Antioxidant activities of cell culture extracts were evaluated111 by the Schaal oven test in sunflower oil and using the DPPH radical. Oxidation was followed by measuring PV. The activity of ethyl acetate extracts was comparable to that of caffeic acid and greater than that of BHT. Extracts and caffeic acid were much stronger scavengers of DPPH free radical than BHT on an equimolar basis. This raises the question as to whether results should be expressed on a mass or equimolar basis. Hydroxycinnamic acids are an important group of antioxidants and their antioxidant and free radical scavenging activities were measured112 by Rancimat and the DPPH radical assay. A number of differences in activity were observed between the two systems and depending on whether lard or corn oil was used in the Rancimat.
The oxidative stability of lard and tallow was examined113 with and without antioxidants by four accelated stability tests. The results suggested that the Rancimat may be the least reliable method. However, it was recommended that more than one accelerated stability test should be used to determine antioxidant effectiveness. A flow injection procedure using amperometric detection of oxidizable substrate (e.g., α-tocopherol plus phenolics) has been proposed114 as an alternative to Rancimat and ABTS radical tests for the evaluation of antioxidant activity of olive oils. Advantages claimed for the proposed procedure are that it is based on the chemical structure of the antioxidant and does not involve accelerated test conditions.
5.2 Peroxide value
This parameter represents the total hydroperoxide and peroxide oxygen content of lipids or lipid-containing materials. It is commonly calculated from an iodometric titration developed over 60 years ago115 that is the basis of current standard methods116,117 for determining PV. In this method hydroperoxides and peroxides oxidize aqueous iodide to iodine which is then titrated with standard thiosulfate solution and starch as end-point indicator. The reactions and stoichiometries for this method are| ROOH + 2H+ +2I− → I2 + ROH + H2O |
| ROOR + 2H+ +2I− → I2 + 2ROH |
| I2 + 2S2O32− → S4O62− + 2I− |
As hydroperoxides are the primary products of lipid oxidation and play a central role in the further autoxidation of lipids, the inhibition of formation and/or action of these unstable species by antioxidants can be used44,120 as a means of assessing antioxidant activity. For example, antioxidant activities of sage, sweet grass and camomile were tested121 in rapeseed oil at 40 °C. Peroxide value, induction period (defined as the time when the PV reached 20 mequiv. kg−1) and protection factors were measured and used to assign relative activities to the extracts. Linoleic acid and antioxidant122–124 were incubated at 40–50 °C for 7 d in the dark, following which time the hydroperoxides from linoleic acid oxidation were determined56 by the iron thiocyanate method. Antioxidant activity was expressed as a reduction in oxidation relative to a control (untreated) sample. Using this approach, the relative antioxidant activities of lime peel fibre and orange peel fibre123 were determined. A limitation in this approach is that hydroperoxides are unstable and extensive oxidation of a lipid can occur without an accompanying build-up in hydroperoxides. However, antioxidants may still exert a significant inhibitory action on transient hydroperoxides, but it will simply not be detected by this test procedure. Therefore, it may be necessary to run control samples to establish that hydroperoxide build-up does indeed occur for the substrate and test conditions chosen. The method should, however, be of value in assessing antioxidant activity during the early stages of lipid oxidation under mild conditions.
5.3 Diene conjugation
In 1931, Gillam and co-workers demonstrated that natural fats develop an absorption peak near 230–235 nm on storage.125 Two years later it was discovered that the peak arose from a diene conjugated bond. It was not until the 1960s, however, that monitoring diene conjugation emerged as a useful technique for the study of lipid oxidation. Diene conjugation resulting from lipid oxidation126 is now commonly used as an end-point for determining the antioxidant activity of a sample. The usual substrate for the determination of conjugated dienes includes any substance containing polyunsaturated fatty acids, with oxidation being initiated90,127,128 by the addition of copper ions, iron ions, AAPH or DDPH or the application of heat. Initially, the lipid undergoes hydrogen abstraction from a CH2 group and the product is usually stabilized by a molecular rearrangement to form a conjugated diene. Quantification of the conjugated dienes may be achieved90 by calculating the increase in absorbance per mass of sample at a fixed time. Lag phase measurements and percentage inhibition have also been used129,130 to quantify results. The antioxidant activity of 44 different berry and fruit wines and liquors was compared126 by conjugated diene measurement with methyl linoleate as substrate. Removal of sugars from the samples was a necessary step to prevent interference during oxidation of the methyl linoleate.As early as 1972, DiLuzio showed that there is a considerable amount of diene conjugated material in human serum lipid extracts.125 He suggested that serum diene conjugation might reflect oxidation in vivo. Moreover, 95% of diene conjugation in human serum, tissue fluids and tissues,125 both abnormal and normal, is due to a single fatty acid. The use of HPLC to separate the UV-absorbing ‘diene conjugate’ material from human body fluids revealed131 that most or all of it consisted of an isomer of linoleic acid, octadeca-9(cis),11(trans)-dienoic acid.
The measurement of the formation of diene conjugation has the advantage that it measures an early stage in the oxidation process. However, even in simple lipid systems, diene conjugation by UV spectroscopy is a generic measurement, providing little information about the structure of the compounds. Selectivity can be enhanced by separation of different diene conjugates using HPLC or by matrix subtraction using second-derivative spectroscopy.131 In either case, sensitivity may also be increased.
Diene conjugation measurements often cannot be performed directly on tissues and body fluids because many other interfering substances are present,132 such as haem proteins, chlorophylls, purines and pyrimidines that absorb strongly in the UV region. Extraction of lipids into organic solvents before analysis is a common approach to this problem.
The antioxidant activities of the flavonoids eriocitrin, diosmin, hesperidin and narirutin extracted from lemon fruit were examined122 using a liposome and an LDL oxidation system. In the liposome system, lipid oxidation was induced by AAPH and the extent of inhibition by added antioxidant was determined as TBARS at 532 nm. For the LDL system, the effect of antioxidant on lag time of the copper(II)-mediated oxidative modification of LDL was measured by monitoring conjugated diene formation at 234 nm. Flavonoid glycosides generally exhibited weaker activity than the corresponding aglycones. Eriocitrin exhibited the highest activity of all lemon constituents as measured by all three methods. Its metabolites by intestinal bacteria (the aglycone eriodictyol, 3,4-dihydroxyhydrocinnamic acid and phloroglucinol) exhibited weaker antioxidative activity but nevertheless exhibited greater activity than α-tocopherol in the LDL oxidation system and had approximately the same activity as (-)-epigallocatechin gallate.
Catechins and procyanidins from cocoa were also studied133 in two in vitro systems: liposomes and human LDL. Liposome oxidation (evaluated as TBARS formation) was initiated with AAPH, AMVN or iron/ascorbate and LDL oxidation (evaluated as formation of conjugated dienes) was initiated with Cu2+ or AAPH. When liposome oxidation was initiated in the aqueous phase, monomer, dimer and trimer fractions were the most effective antioxidants. The higher molecular weight procyanidins were the most effective antioxidants when oxidation was initiated in the lipid domains.
5.4 Thiobarbituric acid reactive substances (TBARS) assay
The TBARS assay was proposed over 40 years ago and is now the most commonly used method134 to detect lipid oxidation. This procedure measures the MDA formed as the split product of an endoperoxide of unsaturated fatty acids resulting from oxidation of a lipid substrate. It is postulated that the formation of MDA from fatty acids with less than three double bonds (e.g., linoleic acid) occurs via the secondary oxidation of primary carbonyl compounds (e.g., non-2-enal).135 The MDA is reacted with thiobarbituric acid (TBA) to form a pink pigment (TBARS) (Fig. 1) that is measured spectrophotometrically136 at its absorption maximum at 532–535 nm.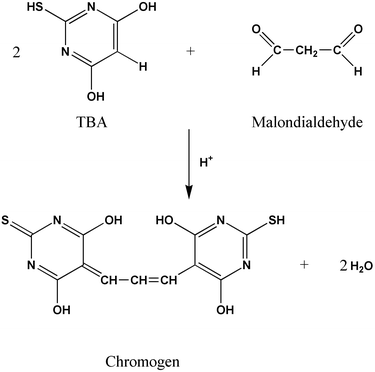 | ||
| Fig. 1 Chromophore formed by condensation of MDA with TBA. | ||
Numerous substrates137–139 have been used in the determination of TBARS, including tissue samples, linoleic and other fatty acids and LDL. A number of model linoleic acid systems have been developed,137,140,141 including emulsions of linoleic acid with SDS or Tween. Ethanol is added to aid in the mixing of the antioxidant with the linoleic acid. The addition of ethanol has recently come under discussion as there is growing evidence142 that ethanol is in itself an antioxidant. Studies by Belguendouz et al.,143 however, found that the presence or absence of ethanol did not influence the antioxidant activity of their samples.
The procedure involves two distinct steps: the substrate is oxidized with the addition of a transition metal ion such as copper or iron or a free radical source such as AAPH (also referred to as ABAP144,145) and then the extent of oxidation is determined by addition of TBA and spectrophotometric measurement of the product. Oxidation is inhibited by the addition of an antioxidant and therefore a reduction in the absorbance is seen. Results are typically quantified146 against a calibration curve for malondialdehyde bis(dimethylacetal) or malondialdehyde bis(diethylacetal), which acts as a source of MDA. Results may also be described4 in terms of percentage inhibition of the oxidation. The TBARS procedure is widely used147,148 even though the reaction is not very specific and reaction conditions have a significant effect on colour development. Selectivity of the TBARS procedure is improved by the use of HPLC to characterize the individual species,149,150 but this still does not identify the source of MDA in samples or eliminate the possibility of a compound with similar spectral properties co-eluting.
Another method for detecting peroxidation in lipids of biological origin151 involves the so-called LPO-586 assay. This method apparently responds to both MDA and 4-hydroxyalkenals but is not specific to either group. The chromophore(s) formed in the condensation of aldehydes with N-methyl-2-phenylindole absorbs strongly close to 586 nm and the method can be used as an alternative to the TBARS method. It has yet to be applied to a wide variety of sample types.
5.5 Measurement of hexanal and related end-products
Decomposition of the primary products of lipid oxidation generates a complex mixture131 including epoxides, ketones (e.g., butanones, pentanones, octanones), hydrocarbons and saturated and unsaturated aldehydes such as hexanal. Various measures of these more or less stable final products of oxidation are used. For instance, anisidine value152 measures 2-alkenals and the oxidation of various oils was followed70,152 by measurement of both anisidine value and PV.The carbonyl compounds including pentanal, deca-2,4-dienal and octa-3,5-dien-2-one are suggested to be the major contributors to off-flavours153–156 associated with the rancidity of many food products. For instance, Fritsch and Gale157 showed that rancid odours occurred in ready-to-eat oat cereals when the hexanal concentration reached 5–10 μg g−1. During rice storage at 40 °C, the appearance of stale flavour158 corresponded to higher levels of propanal, pentanal and hexanal with accompanying decrease in the content of linoleic and linolenic acids. Indeed, the decomposition of the primary oxidation product, 13-hydroperoxide of linoleate ester groups, gives rise to the secondary products which include hexanal, pentane, deca-2,4-dienal and 4-hydroxyalkenals such as 4-hydroxynon-2-enal. Other fatty acid moieties also give rise (via thermolysis of hydroperoxides) to a characteristic set of reaction products159 depending on the mode of oxidation (Table 5).
| Moiety | Autoxidation | Photo-oxidation |
|---|---|---|
| Oleate | Nonanal, octanal | Dec-2-enal |
| Linoleate | Hexanal, pentane, deca-2,4-dienal | Hept-2-enal, hexanal |
| Linolenate | Hepta-2,4-dienal, ethane | Propanal, but-2-enal |
Frankel160 provided a detailed insight into the mechanisms and spectrum of products obtained by lipid autoxidation and such knowledge is useful in recognising the relationship between fatty acid moieties, the intermediate hydroperoxides and the specific volatile secondary metabolites analysed for rancidity or antioxidant studies. Rancidity studies of refined oils and snack foods,161 for example, are frequently based on measuring such secondary oxidation products by headspace GC or GC-MS154,162–164 and correlating these with organoleptic data. Selectivity has been improved165–167 by (isotope dilution) mass spectrometry. More recently, solid-phase microextraction has been applied168 to the determination.
Antioxidant activity can be calculated as percentage inhibition of one or more of the secondary oxidation products relative to a control. The activity of phenolic components of wines75 has been assessed in this manner. Hexanal is the most commonly measured end-product of lipid oxidation169–173 and both sensory and physico-chemical methods165 are used for its determination. Where other antioxidant activity tests may be non-specific, physico-chemical measurement of hexanal174 offers the advantage of analysing a single, well-defined end-product. The significance of hexanal as an analyte for oxidation monitoring (or antioxidant efficiency studies) is indicated by data reported by Snyder et al.175 that show that hexanal formation is usually an order of magnitude higher than with most other secondary oxidation products. An exception is pentane, which forms in concentrations comparable to those of hexanal (pentane formation is an alternative decomposition pathway for 13-hydroperoxide-linoleate). Since pentane is a very stable end-product it may be more suitable than hexanal for monitoring antioxidant activities. Jackson and Giacherio,176 for example, have shown that pentane is one of the main secondary oxidation products formed for soybean oil. In fact, monitoring only one or two analytes may be cyclopian in approach. Several volatile carbonyl compounds were measured177 in human breath following trapping as their 2,4-dinitrophenylhydrazone derivatives. Analysis of the full range of volatile secondary oxidation products (which can easily be done these days by GC-MS) may be the preferred approach.
There is ample evidence178–183 that ethane and pentane (end-products of the oxidation of n - 3 and n - 6 polyunsaturated fatty acids, respectively) in expired air are useful markers of in vivo lipid peroxidation. The major difficulty is contamination from ambient-air ethane and pentane184 and the effective removal of ambient-air hydrocarbons from the subject’s lungs before collection becomes an important step in standardizing the collection procedure. Oxidative stress status was evaluated by breath pentane measurements185 whilst antioxidant status was evaluated by measurement of the total antioxidant capacity of the plasma. These clinical markers of antioxidant and oxidative stress status were not correlated with normal concentrations of carotenoids in plasma and tissues, although vitamin E and β-carotene supplementation186 decreased hydrocarbon excretion.
The quantification of aldehydes such as 4-hydroxynonenal is of great interest not only in that they may indicate levels of autoxidation and hence antioxidant activity but also in that they are extremely reactive and cytoxic. For example, the cytotoxicity of 4-hydroxynonenal is exhibited in diverse processes187,188 such as stimulation of neutrophil chemotaxis and inhibition of many enzymes. This extreme reactivity and metabolic conversion, however, may make them unsuitable as test analytes for in vivo antioxidant activity studies except at high levels of oxidative stress. Furthermore, simple chemical tests such as the TBARS and LPO-586 tests are not specific for this substance. More selective tests based on derivatisation and HPLC, GC or GC-MS189 are more suitable.
The degradation products of oxidation have also been measured indirectly. For instance, the rate of oxidative destruction of β-carotene by degradation products of linoleic acid has been measured190–192 spectrophotometrically at 450–470 nm. An aqueous emulsion of the linoleic acid substrate, carotene and antioxidant were mixed and the results were used to measure antioxidant activity in wines193 and berries.192 Results from the β-carotene procedure were compared192 with MDA production as measured by HPLC and a free radical procedure using DPPH. Results from the various procedures were generally similar. The naturally occurring phenolics showed pro-oxidant activity at low concentrations, unlike the synthetic antioxidants BHA and BHT.
5.6 Measurement of free radicals
Strategies have been developed for measuring the antioxidant activity as the ability to scavenge free radicals generated in aqueous and lipophilic phases. The ability to scavenge specific radicals may be targeted as, for example, hydroxyl radical,50 superoxide radical194 or nitric oxide radical.195 One approach involves95 the generation of a free radical species and direct measurement of its inhibition due to addition of antioxidant(s). Alternatively, the generation of a radical is coupled to oxidation of a substrate, in which case measurement of the inhibitory effect of an antioxidant is based on detection of either the radical or the products of oxidation. For example, the production of peroxyl free radicals by the thermal decomposition of AAPH can be coupled to the oxidation of 2,7-dichlorofluorescin to the fluorescent 2,7-dichlorofluorescein. In this instance, the effect of added antioxidant was seen196 as an increase in the lag phase.The radical that is generated varies and systems have been described using horseradish peroxidase–H2O2,95o-phenylenediamine–H2O2, copper(II)–cumene hydroperoxide, trichloromethyl peroxyl radical,50 DPPH128,197,198 and azo compounds such as the chromogenic redox indicator ABTS.199 End-point detection also varies and has been based on measurement of fluorescence inhibition, chemiluminescence,200,201 oxygen uptake and absorbance.64
Applications which illustrate the potential of spin trapping methods in antioxidant action include the determination of the antioxidant potential of tea extracts in aqueous and organic media,211 assessing the antioxidant contribution of quercetin and other flavanols to the antioxidant capacity of red wines,212 specific assays for the hydroxyl or superoxide radicals in natural extracts or biological systems,210,213,214 the study of free radical transfer in fish lipid–protein systems215 and the measurement of antioxidant capacity from ascorbic acid in blood plasma.216 The specificity, ability to handle complex biological samples and the capacity to identify individual free radicals represent distinct advantages for ESR methods. Nevertheless, applications are limited to date owing mainly to the sensitivity problem. Other problems include the specialist nature and relatively large size and cost of the equipment and that such instrumentation has yet to be developed to the stage where short-lived radicals can be measured in vivo (as NMR imaging has for hydrogen nuclei). Another problem208,209 is that spin traps exhibit widely differing trapping efficiencies for different radicals. Furthermore, spin traps can perturb systems under investigation. For example, it has been shown that such traps can exhibit both oxidant131 and antioxidant217 action, while spin adducts can act as antioxidants.213 Even with these limitations, there is no doubt that ESR will continue to provide valuable information on the complex roles and patterns of free radicals in biological oxidation processes.
Results were expressed by comparison with standard amounts of the synthetic antioxidant trolox (a water-soluble vitamin E analogue) to give rise to the TEAC. The TEAC64,221 is equal to the millimolar concentration of a trolox solution having the antioxidant capacity equivalent to a 1.0 mM solution of the substance under investigation. As used by Rice-Evans and Miller,64 the TEAC reflects the relative ability of hydrogen- or electron-donating antioxidants to scavenge the ABTS radical cation compared with that of Trolox. The ABTS assay has been used93 to measure the total antioxidant activity in pure substances, in body fluids and in plant material. Miller and Rice-Evans199 reported the TEAC of orange and apple juices and blackcurrant drink (Ribena) and also the contribution of individual phenolic antioxidants. The bulk of the TAA of apple juice could be accounted for by chlorogenic acid and the phloretins, whereas in both orange juice and Ribena, vitamin C was the major antioxidant. However, in the case of orange juice, HPLC required preliminary filtration and the measured composition reflected the soluble222 flavonoid portion only. The authors concluded that the phenolic antioxidants protected vitamin C against oxidative decomposition, with those in blackcurrant having the greatest vitamin C-sparing activity. However, the situation is complex and winemakers add ascorbic acid during fermentation as an anti-browning agent, presumably to protect the phenolics against oxidation. TEAC assays have also been measured for flavonol and catechin metabolites as the antioxidant capacities of such metabolites may be significantly different to that of the original antioxidant223 for in vivo processes.
The method of Arnao et al.95 is similar to that of Rice-Evans and Miller64 but differs in a number of important aspects. Unlike the latter method that used the metmyoglobin peroxidase activity, a commercial peroxidase was used by Arnao et al. Arnao et al.95 reported no interferences at the optimal wavelength of 414 nm and this translated to better detection limits. The TAA of orange and grapefruit juices95 were 4.3 and 6.1 mM L−1 ascorbic acid equivalents, respectively.
ROS and DPPH scavenging abilities of extracts of evening primrose227 and citrus essential oils228 have been studied. Citrus oils were examined228 by HPLC using DPPH and results expressed in Trolox equivalents. Plant extracts were separated229 by HPLC and reacted post-column with DPPH and the bleaching was detected as a negative peak by an absorbance detector at 517 nm. Coulometric detection has also been used230 for phenolic plant extracts. A relationship between potential and DPPH scavenging was observed for phenolic acids but not for flavonoids.
The molecular mechanisms and radical scavenging activities of (+)-catechin, ethyl gallate, ascorbic acid and α-tocopherol for DPPH were studied231 by 13C NMR. (+)-Catechin reacted with DPPH to form an o-quinone structure in the B-ring. Phenolic compounds generally exhibited significant scavenging effects against the DPPH free radical.86,190,232–234 DPPH reduction has been compared with other methods including the ABTS assay,235 superoxide-anion scavenging and lipid oxidation.236,237 The antioxidant activity of pomegranate juices was evaluated235 by DPPH and ABTS and the results were compared with those of red wine and tea infusions. Hydrolysable tannins accounted for the high activity of juices. The antioxidant activity of plant biophenols has been attributed238 to trapping of ROS and regeneration of endogenous membrane-bound α-tocopherol. The phenols form o-quinone intermediates upon H-atom abstraction from DPPH and subsequent radical disproportionation. The course of subsequent reactions was dependent on the nature of the phenol, although formation of a dimer239 was a common occurrence.
It is worth reiterating that the ABTS and DPPH methods are substrate-free. Their popularity can be attributed to simplicity and speed of analysis, but this is achieved at a potential price and the relevance of data generated with these procedures must be considered carefully.
5.7 Other measures of antioxidant activity
Phycoerythrin is also used to assess the effectiveness of antioxidants against hydroxyl radicals. Hydroxyl radicals are generated from an ascorbate–Cu2+ system at copper-binding sites on macromolecules. Site specific damage to macromolecules results from the reaction
| Target–Cu2+ + HO• → damaged target + Cu2+ |
This assay is particularly useful in screening for compounds that protect against damage by chelating metal ions necessary for site-specific formation of the radical species. The inhibition of oxidation by an antioxidant can be examined by the retardation of the loss of fluorescence, with the inhibition being proportional to the antioxidant activity. Final results can be calculated244–246 using the differences in areas under the phycoerythrin decay curves between the blank and a sample and are expressed in trolox equivalents.
Antioxidant activities of several juices and fruits were reported244,247 as the automated oxygen radical absorbance capacity (ORAC) in micromoles of Trolox equivalents. This value combined both inhibition time and the extent of inhibition into a single quantity244 whereas other methods use either the inhibition time at a fixed inhibition degree or the inhibition degree at a fixed time as the basis for quantifying results. There was significant variation in the TAA of several fruits with strawberry having the highest ORAC activity on the basis of both wet and dry weight of fruit. The contribution of vitamin C to the activity was <15% except for kiwi fruit and honey dew melon. Most of the antioxidant capacity of these fruits was from the juice fractions. The contribution of the fruit pulp fraction (extracted with acetone) to the total ORAC activity of a fruit was usually <10%.
ORAC values showed a significant positive linear correlation246 with electrochemical data obtained by HPLC with coulometric array detection. Phenolic acids, in general, had lower antioxidant activities against peroxyl radicals than flavonoids that contained multiple hydroxyl groups. However, the flavonoid glycosides (including rutin, naringin and hesperidin) usually had low ORAC activities. A number of factors determine antioxidant activity including reactivity as a hydrogen- or electron-donating agent and this aspect relates to its reduction potential. Indeed, there is broad agreement92 between the half-peak reduction potential and the TAA as measured by TEAC. This was rationalized on the basis that both electrochemical oxidation and hydrogen-donating free radical scavenging involve the rupture of the same phenolic bond. Thus, with the exception of kaempferol, flavonoids with efficient scavenging properties had a TEAC value exceeding 1.9 mM and a half-peak reduction potential below 0.2 mV. This correlation may be fortuitous as the half-peak reduction potentials are thermodynamically meaningless unless the electrochemical processes are reversible, a condition that is seldom valid.
Results can be standardized by addition of Trolox to the sample after consumption of natural antioxidants to produce a second induction period. Stoichiometric factors for pure antioxidants are different218 (e.g., Trolox, 2.0; ascorbate, 1.5; urate, 1.7) and these must be taken into account when extrapolating results back to molar concentrations from TRAP values. The method is time consuming and suffers a number of problems,64,250 although the concept has been very useful for quantifying and comparing251 antioxidant capacity.
The antioxidant activity of four standard antioxidants (gallic acid, uric acid, Trolox and ascorbic acid) was compared252 using TEAC and TRAP assays and LDL oxidation. The results were not comparable in that gallic acid was the strongest antioxidant in all three systems but the relative activity of the remaining compounds depended on the system.
Three different methods were also used253 for quantifying the antioxidant capacity of LDL ex vivo in dyslipidaemic patients with coronary heart disease. These involved determination of LDL TRAP in plasma AMVN-induced oxidation and measuring the extinction time of chemiluminescence, conjugated diene formation in copper-induced oxidation and consumption times of reduced α-tocopherol and ubiquinol in AMVN-induced oxidation. Tocopherol supplementation produced statistically significant changes in all antioxidant variables except those related to LDL ubiquinol. It was concluded that LDL TRAP assay may complement the other methods used to quantify the antioxidant capacity of LDL.
Although phenols exert strong antioxidant activity, in vivo evidence254 has produced contradictory results. When ingested by healthy volunteers, red wine and green tea were the most efficient in protecting LDL from oxidation driven by peroxyl and ferryl radicals, respectively. However, the phenolic content alone was not an index of their in vivo antioxidant activity. Moreover, certain phenols such as quercetin have a biphasic effect255 depending on dose. The beneficial effect of natural and synthetic antioxidants on surrogate markers of vascular disease such as endothelial function and LDL oxidation have been demonstrated. Antioxidant activity in various substrates and tests including LDL in vitro is related256 to the molarity of wine or juice phenolics. Data are limited but the concentrations of the major dietary phenols may be substantially lower than those seen to be effective in in vitro test systems. However, it is very difficult to extrapolate meaningfully to the human in vivo situation because of uncertainties about absorption and pharmacokinetics.257 The antioxidants, uric acid and serum albumins are present in considerably greater molar concentrations than the metabolites of dietary phenols. Furthermore, no beneficial effect has been demonstrated258 upon vascular mortality in high-risk individuals in large prospective randomized controlled intervention trials. The pro-oxidant effects of high dose antioxidant supplements, particularly in patients with established vascular disease, may have contributed to these results.
6 Summary
Antioxidant activity has been assessed in many ways. The limitation of many newer methods is the frequent lack of an actual substrate in the procedure. The combination of all approaches with the many test methods available explains the large variety of ways in which results of antioxidant testing are reported. The measurement of antioxidant activities, especially of antioxidants that are mixtures, multifunctional or are acting in complex multiphase systems, cannot be evaluated satisfactorily by a simple antioxidant test without due regard to the many variables influencing the results. Several test procedures may be required to evaluate such antioxidant activities. A general method of reporting antioxidant activity independent of the test procedure is proposed.References
- V. R. Winrow, P. G. Winyard, C. J. Morris and D. R. Blake, Br. Med. Bull., 1993, 49, 506 Search PubMed.
- V. Bauer, R. Sotnikova, J. Machova, S. Matyas, V. Pucovsky and M. Stefek, Life Sci., 1999, 65, 1909 CrossRef CAS.
- G. U. Bae, D. W. Seo, H. K. Kwon, H. Y. Lee, S. Hong, Z. W. Lee, K. S. Ha, H. W. Lee and J. W. Han, J. Biol. Chem., 1999, 274, 32596 CrossRef CAS.
- L. B. Colbert and E. A. Decker, J. Food Sci., 1991, 56, 1248 Search PubMed.
- F. Shahidi, P. K. Janitha and P. D. Wanasundara, Crit. Rev. Food Sci. Nutr., 1992, 32, 67 Search PubMed.
- M. V. Martinez and J. R. Whitaker, Trends Food Sci. Technol., 1995, 6, 195 CrossRef CAS.
- M. G. Lindley, Trends Food Sci. Technol., 1998, 9, 336 CrossRef CAS.
- S. Beutner, B. Bloedorn, S. Frixel, I. Hernandez Blanco, T. Hoffmann, H-D. Martin, B. Mayer, P. Noack, C. Ruck, M. Schmidt, I. Schulke, S. Sell, H. Ernst, S. Haremza, G. Seybold, H. Sies, W. Stahl and R. Walsh, J. Sci. Food Agric., 2001, 81, 559 CrossRef CAS.
- P. C. H. Hollman and I. C. W. Arts, J. Sci. Food Agric., 2000, 80, 1081 CrossRef CAS.
- M. Burits, K. Asres and F. Bucar, Phytother. Res., 2001, 15, 103 CrossRef CAS.
- F. Shahidi, U. D. Chavan, M. Naczk and R. Amarowicz, J. Agric. Food Chem., 2001, 49, 926 CrossRef CAS.
- K. Sakata, in Food and Free Radicals, ed. M. Hiramatsu, T. Yoshikawa and M. Inoue, Plenum Press, New York, 1997, pp. 85–99 Search PubMed.
- E. Miro-Casas, M. F. Albaladejo, M. I. Covas, J. O. Rodriguez, E. M. Colomer, R. M. L. Raventos and R. de la Torre, Anal. Biochem., 2001, 294, 63 CrossRef CAS.
- M. B. Arnao, Trends Food Sci. Technol., 2001, 11, 419 Search PubMed.
- A. N. Glazer, Methods Enzymol., 1990, 186, 161 Search PubMed.
- S. McDonald, P. D. Prenzler, M. Antolovich and K. Robards, Food Chem., 2001, 73, 73 CrossRef CAS.
- J. C. Espin, C. Soler-Rivas and H. J. Wichers, J. Agric. Food Chem., 2000, 48, 648 CrossRef CAS.
- E. N. Frankel and A. S. Meyer, J. Sci. Food Agric., 2000, 80, 1925 CrossRef CAS.
- Y. Ming-Hua and K. M. Schaichx, Free Radical Biol. Med., 2000, 20, 225 CrossRef.
- G. O. Adegoke, M. V. Kumar, A. G. G. Krishna, M. C. Varadaraj, K. Sambaiah and B. R. Lokesh, J. Food Sci. Technol. Mysore, 1998, 35, 283 Search PubMed.
- G. Lercker, R. Bortolomeazzi and L. Pizzale, J. Am. Oil Chem. Soc., 1998, 75, 1115 Search PubMed.
- C. Tanielian and R. Mechin, Photochem. Photobiol., 1994, 59, 263 Search PubMed.
- K. F. Gey, Bibl. Nutr. Dieta, 1986, 37, 53 Search PubMed.
- R. J. Hamilton, in Rancidity in Foods, ed. J. C. Allen and R. J. Hamilton, Applied Science, London, 1983, pp. 1–20 Search PubMed.
- J. Kanner, J. B. German and J. E. Kinsella, Crit. Rev. Food Sci. Nutr., 1987, 25, 317 Search PubMed.
- K. H. Cheeseman and T. F. Slater, Br. Med. Bull., 1993, 49, 481 Search PubMed.
- D. J. McClements and E. A. Decker, J. Food Sci., 2000, 65, 1270 Search PubMed.
- K. Warner and S. Knowlton, J. Am. Oil Chem. Soc., 1997, 74, 1317 Search PubMed.
- O. O. Lasekan, W. Lasekan, M. A. Idowu and O. A. Ojo, J. Cereal Sci., 1996, 24, 79 CrossRef.
- J. H. Nielsen, C. E. Olsen, J. Lyndon, J. Sorensen and L. H. Skibsted, J. Dairy Res., 1996, 63, 615 Search PubMed.
- D. N. R. Rao and A. I. Cederbaum, Free Radical Biol. Med., 1997, 22, 439 CrossRef CAS.
- C. K. B. Ferrari, Biologia, 2000, 55, 581 Search PubMed.
- M. T. Corasaniti, M. C. Strongoli, D. Rotiroti, G. Bagetta and G. Nistico, Pharmacol. Toxicol., 1998, 83, 1 Search PubMed.
- R. Baliga, N. Ueda, P. D. Walker and S. V. Shah, Drug Metab. Dispos. Rev., 1999, 31, 971 Search PubMed.
- J. M. C. Gutteridge, Chem.-Biol. Interact., 1994, 91, 133 Search PubMed.
- S. J. Jadhav, S. S. Nimbalkar, A. D. Kulkarni and D. L. Madhavi, in Food Antioxidants: Technological, Toxicological and Health Perspectives, ed. D. L. Madhavi, S. S. Deshpande and D. K. Salunkhe, Marcel Dekker, New York, 1996, pp. 5–64 Search PubMed.
- C. Leclercq, D. Arcella and A. Turrini, Food Chem. Toxicol., 2000, 38, 1075 CrossRef CAS.
- G. C. Maziero, C. Baunwart, M. Cecilia and F. Toledo, Braz. Food Addit. Contam., 2001, 18, 365 Search PubMed.
- F. Bonilla, M. Mayen, J. Merida and M. Medina, Food Chem., 1999, 66, 209 CrossRef CAS.
- F. Iverson, Food Chem. Toxicol., 1999, 37, 993 CrossRef CAS.
- G. M. Williams, M. J. Iatropoulos and J. Whysner, Food Chem. Toxicol., 1999, 37, 1027 CrossRef CAS.
- Y. S. Velioglu, G. Mazza, L. Gao and B. D. Oomah, J. Agric. Food Chem., 1998, 46, 4113 CrossRef CAS.
- P. M. Clarkson, Crit. Rev. Food Sci. Nutr., 1995, 35, 131 Search PubMed.
- Y. B. C. Man and C. P. Tan, J. Am. Oil Chem. Soc., 1999, 76, 331 Search PubMed.
- C. Rice-Evans, D. Leake, K. R. Bruckdorfer and A. T. Diplock, Free Rad. Res., 1996, 25, 285 Search PubMed.
- J. T. Salonen, Free Rad. Res., 2000, 33(Suppl S), S41 Search PubMed.
- D. Pratico, J. A. Lawson, J. Rokach and G. A. FitzGerald, Trends Endocrinol. Metab., 2001, 12, 243 CrossRef CAS.
- C. Sanchez-Moreno, A. Jimenez-Escrig and F. Saura-Calixto, Nutr. Res., 2000, 20, 941 CrossRef CAS.
- F. Visioli, D. Caruso, C. Galli, S. Viappiani, G. Galli and A. Sala, Biochem. Biophys. Res. Commun., 2000, 278, 797 CrossRef CAS.
- O. I. Aruoma, J. P. E. Spencer, D. Warren, P. Jenner, J. Butler and B. Halliwell, Food Chem., 1997, 60, 149 CrossRef CAS.
- C. Sanbongi, N. Osakabe, M. Natsume, T. Takizawa, S. Gomi and T. Osawa, J. Agric. Food Chem., 1998, 46, 454 CrossRef CAS.
- V. W. Bowry and R. Stocker, J. Am. Chem. Soc., 1993, 115, 6029 CrossRef CAS.
- J. N. Chacon, P. Gaggini, R. S. Sinclair and F. J. Smith, Chem. Phys. Lipids, 2000, 107, 107 CrossRef CAS.
- Q. T. Li, M. H. Yee and B. K. Tan, Biochem. Biophys. Res. Commun., 2000, 273, 72 CrossRef CAS.
- S. Z. Dziedzic and B. J. F. Hudson, J. Am. Oil Chem. Soc., 1984, 61, 1042 Search PubMed.
- E. M. Marinova and N. Yanishlieva, Food Chem., 1996, 56, 139 CrossRef CAS.
- D. Mantle, J. G. Anderton, G. Falkous, M. Barnes, P. Jones and E. K. Perry, Comp. Biochem. Physiol., B: Biochem. Mol. Biol., 1998, 121, 385 Search PubMed.
- K. Schwarz, G. Bertelsen, L. R. Nissen, P. T. Gardner, M. I. Heinonen, A. Hopia, T. Huynh-Ba, P. Lambelet, D. McPhail, L. H. Skibsted and L. Tijburg, Eur. Food Res. Technol., 2001, 212, 319 Search PubMed.
- S. Z. Dziedzic and B. J. F. Hudson, Food Chem., 1983, 11, 161 CrossRef CAS.
- R. J. Hamilton, C. Kalu, E. Prisk, F. B. Padley and H. Pierce, Food Chem., 1997, 60, 193 CrossRef CAS.
- T. Nishiike, S. Kondo, T. Yamamoto, A. Shigeeda, Y. Yamamoto, H. Takamura and T. Matoba, Biosci. Biotechnol. Biochem., 1997, 61, 1973 Search PubMed.
- M. T. Satué-Gracia, M. Heinonen and E. N. Frankel, J. Agric. Food Chem., 1997, 45, 3362 CrossRef CAS.
- N. Salah, N. J. Miller, G. Paganga, L. Tijburg, G. P. Bolwell and C. Rice-Evans, Arch. Biochem. Biophys., 1995, 322, 339 CrossRef CAS.
- C. E. Rice-Evans and N. J. Miller, Methods Enzymol., 1994, 234, 279 Search PubMed.
- R. A. Garcia, J. H. Hotchkiss and K. H. Steinkraus, Food Addit. Contam., 1999, 16, 63 CrossRef CAS.
- A. I. Hopia, S. W. Huang, K. Schwarz, J. B. German and E. N. Frankel, J. Agric. Food Chem., 1996, 44, 2030 CrossRef CAS.
- M. Watanabe, J. Agric. Food Chem., 1998, 46, 839 CrossRef CAS.
- D. Bonnefont-Rousselot, A. Rouscilles, C. Bizard, J. Delattre, D. Jore and M. Gardes-Albert, Radiat. Res., 2001, 155, 279 Search PubMed.
- N. Gamez-Meza, J. A. Noriega-Rodriguez, L. A. Medina-Juarez, J. Ortega-Garcia, R. Cazarez-Casanova and O. Angulo-Guerrero, J. Am. Oil Chem. Soc., 1999, 76, 1445 Search PubMed.
- A. M. Lampi, A. Hopia and V. Piironen, J. Am. Oil Chem. Soc., 1997, 74, 549 Search PubMed.
- N. Nakatani, Y. Tachibana and H. Kikuzaki, J. Am. Oil Chem. Soc., 2001, 78, 19 Search PubMed.
- A. Bocco, M. E. Cuvelier, H. Richardand and C. Berset, J. Agric. Food Chem., 1998, 46, 2123 CrossRef CAS.
- A. Bocco, M. E. Cuvelier, H. Richard and C. Berset, Sci. Aliment., 1998, 18, 13 Search PubMed.
- E. N. Frankel, J. B. German and P. A. Davis, Lipids, 1992, 27, 1047 Search PubMed.
- E. N. Frankel, A. L. Waterhouse and P. L. Teissedre, J. Agric. Food Chem., 1995, 43, 890 CrossRef CAS.
- S. Miura, J. Watanabe, M. Sano, T. Tomita, T. Osawa, Y. Hara and I. Tomita, Biol. Pharm. Bull., 1995, 18, 1 Search PubMed.
- D. Harats, S. Chevion, M. Nahir, Y. Norman, O. Sagee and E. M. Berry, Am. J. Clin. Nutr., 1998, 67, 240 Search PubMed.
- A. Ghiselli, A. Damicis and A. Giacosa, Eur. J. Cancer Prev., 1997, 6, 15.
- T. F. Slater, Am. J. Clin. Nutr., 1991, 53, 394S Search PubMed.
- M. Heinonen, D. Rein, M. T. Satue Gracia, S. W. Huang, J. B. German and E. N. Frankel, J. Agric. Food Chem., 1998, 46, 917 CrossRef CAS.
- E. Lovaas, J. K. Midtbo and T. Strom, Pathophysiology, 1998, 5, 84 CrossRef.
- A. Arora, M. G. Nair and G. M. Strasburg, Free Rad. Biol. Med., 1998, 24, 1355 CrossRef CAS.
- W. Bors, C. Michel and K. Stettmaier, Methods Enzymol., 2001, 335, 166 Search PubMed.
- W. F. Hodnick, F. S. Kung, W. J. Roettger, W. C. Bohmont and R. S. Pardini, Biochem. Pharmacol., 1986, 35, 2345 CrossRef CAS.
- G. W. Plumb, K. R. Price, M. T. Garcia-Conesa and G. Williamson, presented at Polyphenols Communications 98, XIXth International Conference on Polyphenols, Lille, 1–4 September 1998.
- F. A. M. Silva, F. Borges, C. Guimaraes, J. L. F. C. Lima, C. Matos and S. Reis, J. Agric. Food Chem., 2000, 48, 2122 CrossRef CAS.
- A. K. Ratty and N. P. Das, Biochem. Med. Metab. Biol., 1988, 39, 69 CrossRef CAS.
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Free Rad. Biol. Med., 1996, 20, 933 CrossRef CAS.
- G. Kansci, C. Genot, A. Meynier and G. Gandemer, Food Chem., 1997, 60, 165 CrossRef CAS.
- E. N. Frankel, S. W. Huang, E. Prior and R. Aeschbach, J. Sci. Food Agric., 1996, 72, 201 CrossRef CAS.
- Y. Yamaguchi, S. Kagota, K. Nakamura, K. Shinozuka and M. Kunitomo, Phytother. Res., 2000, 14, 647 CrossRef CAS.
- K. Robards, P. D. Prenzler, G. Tucker, P. Swatsitang and W. Glover, Food Chem., 1999, 66, 401 CrossRef CAS.
- A. Cano, J. Hernandez-Ruiz, F. Garcia-Canovas, M. Acosta and M. B. Arnao, Phytochem.l Anal., 1998, 9, 196 Search PubMed.
- N. Pellegrini, F. Visioli, S. Buratti and F. Brighenti, J. Agric. Food Chem., 2001, 49, 2532 CrossRef CAS.
- M. B. Arnao, A. Cano, J. Hernández-Ruiz, F. García-Cánovas and M. Acosta, Anal. Biochem., 1996, 236, 255 CrossRef CAS.
- C. Armando, S. Maythe and N. P. Beatriz, J. Sci. Food Agric., 1998, 77, 463 CrossRef CAS.
- P. Zandi and L. Ahmadi, J. Food Sci. Technol. Mysore, 2000, 37, 436 Search PubMed.
- M. Mikula and A. Khayat, J. Am. Oil Chem. Soc., 1985, 62, 1694 Search PubMed.
- Y. Fukuda, T. Osawa, M. Namiki and T. Ozaki, Agric. Biol. Chem., 1985, 49, 301 Search PubMed.
- S. Z. Dziedzic and B. J. F. Hudson, Food Chem., 1983, 12, 205 CrossRef CAS.
- G. C. Yen and C. E. Lee, J. Sci. Food Agric., 1997, 75, 326 CrossRef CAS.
- J. Sanhueza, S. Nieto and A. Valenzuela, J. Am. Oil Chem. Soc., 2000, 77, 933 Search PubMed.
- M. Baldioli, M. Servili, G. Perretti and G. F. Montedoro, J. Am. Oil Chem. Soc., 1996, 73, 1589 Search PubMed.
- A. Castera-Rossignol and F. Bosque, Corps Gras Lipides, 1994, 1, 131 Search PubMed.
- G. Papadopoulos and D. Boskou, J. Am. Oil Chem. Soc., 1991, 68, 669 Search PubMed.
- M. T. Satué, S.-W. Huang and E. N. Frankel, J. Am. Oil Chem. Soc., 1995, 72, 1131 Search PubMed.
- E. N. Frankel, Trends Food Sci. Technol., 1993, 4, 220 CrossRef CAS.
- M. Milovanovic, K. Picuricjovanovic, B. Vucelicradovic and Z. Vrbaski, J. Am. Oil Chem. Soc., 1996, 73, 773 Search PubMed.
- L. A. Quinn and H. H. Tang, J. Am. Oil Chem. Soc., 1996, 73, 1585 Search PubMed.
- R. Aparicio L. Roda M. A. Albi and F. Gutierrez, J. Agric. Food Chem., 1999, 47, 4150 CrossRef.
- E. G. Kovatcheva, I. I. Koleva, M. Ilieva, A. Pavlov, M. Mincheva and M. Konushlieva, Food Chem., 2001, 72, 295 CrossRef CAS.
- J. H. Chen and C.-T. Ho, J. Agric. Food Chem., 1997, 45, 2374 CrossRef CAS.
- C. Liang and K. Schwarzer, J. Am. Oil Chem. Soc., 1998, 75, 1441 Search PubMed.
- S. Mannino, S. Buratti, M. S. Cosio and N. Pellegrini, Analyst, 1999, 124, 1115 RSC.
- K. Williams , Oils, Fats and Fatty Foods – Their Practical Examination, Churchill, London, 1966 Search PubMed[p. 53 refers to a special report No. 46 by Lea, dated 1938, referring to PV determination by iodimetry; the procedure, however, had probably been developed much earlier].
- C. Paquot, IUPAC Standard Methods for the Analysis of Oils, Fats and Derivatives, Pergamon Press, Oxford, 6th edn., 1979 Search PubMed.
- Official Methods and Recommended Practices of the American Oil Chemist’s Society, ed. D. Firestone, AOCS Press, Champaign, IL, 4th edn., 1990, Method No. Cd 8-53 Search PubMed.
- F. Shahidi, in Natural Antioxidants – Chemistry, Health Effects and Applications, ed. F. Shahidi, AOCS Press, Champaigh, IL, 1997, ch. 24 Search PubMed.
- F. R. Vandevoort, A. A. Ismail, J. Sedman, J. Dubois and T. Nicodemo, J. Am. Oil Chem. Soc., 1994, 71, 921 Search PubMed.
- I. Jaswir, Y. B. C. Man and D. D. Kitts, J. Am. Oil Chem. Soc., 2000, 77, 1161 Search PubMed.
- D. Bandoniene, A. Pukalskas, P. R. Venskutonis and D. Gruzdiene, Food Res. Int., 2000, 33, 785 CrossRef CAS.
- Y. Miyake, K. Yamamoto, Y. Morimitsu and T. Osawa, J. Agric. Food Chem., 1997, 45, 4619 CrossRef CAS.
- J. A. Larrauri, P. Ruperez, L. Bravo and F. Calixto, Food Res. Int., 1996, 29, 757 CrossRef CAS.
- J. A. Larrauri, P. Ruperez and F. S. Calixto, J. Agric. Food Chem., 1997, 45, 4028 CrossRef CAS.
- T. L. Dormandy and D. G. Wickens, Chem. Phys. Lipids, 1987, 45, 353 CrossRef CAS.
- I. M. Heinonen, P. J. Lehtonen and A. I. Hopia, J. Agric. Food Chem., 1998, 46, 25 CrossRef CAS.
- I. Karmansky and N. Gruener, Clin. Chim. Acta, 1997, 259, 177 CrossRef CAS.
- M. Ohnishi, H. Morishita, H. Iwahashi, S. Toda, Y. Shirataki, M. Kimura and R. Kido, Phytochemistry, 1994, 36, 579 CrossRef CAS.
- J. J. M. Van den Berg, N. E. Cook and D. L. Tribble, Lipids, 1995, 30, 599 Search PubMed.
- S. A. Wiseman, J. N. N. J. Mathot, N. J. Defouw and L. B. M. Tijburg, Atherosclerosis, 1996, 120, 15 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, New York, 3rd edn., 1999 Search PubMed.
- D. L. Madhavi, S. S. Deshpande and D. K. Salunkhe, Food Antioxidants: Technological, Toxicological and Health Perspectives, Marcel Dekker, New York, 1996 Search PubMed.
- S. B. Lotito, L. Actis-Goretta, M. L. Renart, M. Caligiuri, D. Rein, H. H. Schmitz, F. M. Steinberg, C. L. Keen and C. G. Fraga, Biochem. Biophys. Res. Commun., 2000, 276, 945 CrossRef CAS.
- E. Kishida, S. Tokumaru, Y. Ishitani, M. Yamamoto, M. Oribe, H. Iguchi and S. Kojo, J. Agric. Food Chem., 1993, 41, 1598 CrossRef CAS.
- J. Fernández, J. A. Perez-Alvarez and J. A. Fernández-Lopez, Food Chem., 1997, 59, 345 CrossRef CAS.
- R. Guillensans and M. Guzmanchozas, Crit. Rev. Food Sci. Nutr., 1998, 38, 315 Search PubMed.
- Y. Miyake, K. Yamamoto and T. Osawa, J. Agric. Food Chem., 1997, 45, 3738 CrossRef CAS.
- C. Saccini, M. Nardini, M. D’Aquino, V. Gentili, M. Di Felice and G. Tomassi, J. Lipid Res., 1992, 33, 627 Search PubMed.
- K. D. Croft, P. Williams, S. Dimmitt, R. Abu-Amsha and L. Beilin, Biochim. Biophys. Acta, 1995, 1254, 250 CrossRef CAS.
- A. N. Wijewickreme and D. D. Kitts, J. Agric. Food Chem., 1997, 45, 4571 CrossRef CAS.
- T. Osawa and M. Namiki, Agric. Biol. Chem., 1981, 45, 735 Search PubMed.
- M. H. Criqui, BioFactors, 1997, 6, 421 Search PubMed.
- L. Belguendouz, L. Fremont and A. Linard, Biochem. Pharmacol., 1997, 53, 1347 CrossRef CAS.
- M. L. Stecchini, M. Del Torre and M. Munari, Int. J. Food Microbiol., 2001, 64, 183 CrossRef CAS.
- T. Paul, M. J. Young and K. U. Ingold, Biochemistry, 2000, 39, 4129 CrossRef CAS.
- Z. Du and J. Bramlage, J. Agric. Food Chem., 1992, 40, 1566 CrossRef CAS.
- T. Asakawa and S. Matsusshita, Lipids, 1980, 15, 137 Search PubMed.
- T. F. Slater, Methods Enzymol., 1984, 105, 283 Search PubMed.
- S. Chirico, C. Smith, C. Marchant, M. J. Mitchinson and B. Halliwell, Free Rad. Res. Commun., 1993, 19, 51 Search PubMed.
- M. K. Shih and M. L. Hu, Mutat. Res., 1999, 438, 125 CrossRef CAS.
- J. Diaz, E. Serrano, F. Acosta and L. F. Carbonell, Clin. Biochem., 1998, 31, 277 CrossRef CAS.
- F. Z. Sheabar and I. Neeman, J. Am. Oil Chem. Soc., 1988, 65, 990 Search PubMed.
- M. G. Heydanek and R. J. McGorrin, J. Agric. Food Chem., 1981, 29, 1093 CrossRef CAS.
- C. J. Bergman, J. T. Delgado, R. Bryant, C. Grimm, K. R. Cadwallader and B. D. Webb, Cereal Chem., 2000, 77, 454 Search PubMed.
- M. R. Corbo, R. Lanciotti and M. E. Guerzoni, J. Agric. Food Chem., 2000, 48, 2401 CrossRef CAS.
- E. L. Molteberg, E. M. Magnus, J. M. Bjorge and A. Nilsson, Cereal Chem., 1996, 73, 579 Search PubMed.
- C. W. Fritsch and J. A. Gale, J. Am. Oil Chem. Soc., 1977, 54, 225 Search PubMed.
- K. Yasumatsu, S. Moritaka and S. Wada, Agric. Biol. Chem., 1966, 30, 483 Search PubMed.
- K. Warner, in Antioxidant Methodology, ed. O. Aruoma and S. L. Cuppett, AOAC Press, Champaign, IL, 1997, pp. 223–233 Search PubMed.
- E. N. Frankel, J. Am. Oil Chem. Soc., 1985, 61, 1908 Search PubMed.
- K. Robards, A. F. Kerr and E. Patsalides, Analyst, 1988, 113, 213 RSC.
- Y. Seto, J. Chromatogr., 1994, 674, 25 CrossRef CAS.
- A. C. Bovell-Benjamin, L. H. Allen, E. N. Frankel and J. X. Guinard, J. Food Sci., 1999, 64, 371 Search PubMed.
- F. Ulberth and D. Roubicek, Int. Dairy J., 1995, 5, 523 CrossRef CAS.
- K. Robards, A. F. Kerr, E. Patsalides and J. Korth, J. Am. Oil Chem. Soc., 1988, 65, 1621 Search PubMed.
- B. A. Bruenner, A. D. Jones and J. B. German, Anal Biochem., 1996, 241, 212 CrossRef CAS.
- F. St-Germain, B. Vachon, J. Montgomery and C. Des Rosiers, Free Rad. Biol. Med., 1997, 23, 166 CrossRef CAS.
- N. P. Brunton, D. A. Cronin, F. J. Monahan and R. Durcan, Food Chem., 2000, 68, 339 CrossRef CAS.
- L. L. De Zwart, J. Venhorst, M. Groot, J. N. M. Commandeur, R. C. A. Hermanns, J. H. M. Meerman, B. L. M. Van Baar and N. P. E. Vermeulen, J. Chromatogr., B: Biomed. Appl., 1997, 694, 277 Search PubMed.
- C. M. Koelsch, T. W. Downes and T. P. Labuza, J. Food Sci., 1991, 56, 816 Search PubMed.
- H. Takamura, K. Kitamura and M. Kito, FEBS Lett., 1991, 292, 42 CrossRef CAS.
- K. Umegaki, M. Sano and T. Esashi, BoneMarrow Transplant., 1999, 23, 173 CrossRef CAS.
- K. Yoshino, M. Sano and M. Hagiwara, Biol. Pharm. Bull., 1993, 16, 84 Search PubMed.
- P. L. Teissedre, A. L. Waterhouse, R. L. Walzem, J. B. German, E. N. Frankel, S. E. Ebeler and A. J. Clifford, Bull. OIV, 1996, 69, 251 Search PubMed.
- J. M. Snyder, E. N. Frankel and E. Selke, J. Am. Oil Chem. Soc., 1985, 62, 1675 Search PubMed.
- H. W. Jackson and D. J. Giacherio, J. Am. Oil Chem. Soc., 1977, 54, 458 Search PubMed.
- Y. M. Liu, S. R. Dueker, A. D. Jones, S. E. Ebeler and A. J. Clifford, Clin. Chem., 1995, 41, 1028 Search PubMed.
- R. Guilbaud, A. C. Ricard, C. Daniel, S. Boileau, H. Vantra and G. Chevalier, Toxicol. Methods, 1994, 4, 1 CAS.
- M. D. Knutson and F. E. Viteri, Anal. Biochem., 1996, 242, 129 CrossRef CAS.
- E. R. Mohler, P. Reaven, J. E. Stegner, N. S. Fineberg and D. R. Hathaway, J. Chromatogr., B: Biomed. Appl., 1996, 685, 201 Search PubMed.
- A. C. Ricard, . Guilbaud, S. Boileau, H. V. Tra and G. Chevalier, Toxicol. Lett., 1994, 71, 131 CrossRef CAS.
- GM. D. Schweich, D. Lison and R. Lauwerys, Biochem. Pharmacol., 1994, 47, 1395 CrossRef.
- Y. K. Shin, J. V. Collea, Y. D. Kim and S. Y. Kim, Int. J. Obstet Anesth., 1997, 6, 82 CrossRef CAS.
- M. D. Knutson, G. J. Handelman and F. E. Viteri, Free Rad. Biol. Med., 2000, 28, 514 CrossRef CAS.
- P. Borel, P. Grolier, Y. Boirie, L. Simonet, E. Verdier, Y. Rochette, M. C. Alexandregouabau, B. Beaufrere, D. Lairon and V. Azaisbraesco, J. Lab. Clin. Med., 1998, 132, 61 CrossRef CAS.
- C. M. F. Kneepkens, G. Lepage and C. C. Roy, Free Rad. Biol. Med., 1994, 17, 127 CrossRef CAS.
- A. Chandra and S. Srivastra, Lipids, 1997, 32, 779 Search PubMed.
- W. G. Siems, H. Zollner, T. Grune and H. Esterbauer, J. Lipid Res., 1997, 38, 612 Search PubMed.
- M. Kinter, in Free Radicals, a Practical Approach, ed. N. A. Punchard and F. J. Kelly, IRL Press, Oxford, 1996, pp.133–145 Search PubMed.
- R. Amarowicz, M. Naczk and F. Shahidi, J. Agric. Food Chem., 2000, 48, 2755 CrossRef CAS.
- M. S. Taga, E. E. Miller and D. E. Pratt, J. Am. Oil Chem. Soc., 1984, 61, 928 Search PubMed.
- L. R. Fukumoto and G. Mazza, J. Agric. Food Chem., 2000, 48, 3597 CrossRef CAS.
- J. Kanner, E. Frankel, R. Granit, B. German and J. E. Kinsella, J. Agric. Food Chem., 1994, 42, 64 CrossRef CAS.
- Y. H. Chu, C. L. Chang and H. F. Hsu, J. Agric. Food Chem., 2000, 80, 561 CrossRef CAS.
- S. A. B. E. van Acker, M. N. Tromp, G. R. Haenen, W. J. van der Vijgh and A. Bast, Biochem. Biophys. Res. Commun., 1995, 214, 755 CrossRef CAS.
- M. Valkonen and T. Kuusi, J. Lipid Res., 1997, 38, 823 Search PubMed.
- V. Bondet, W. Brand-Williams and C. Berset, Lebensm.-Wiss. Technol., 1997, 30, 609 Search PubMed.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, Lebensm.-Wiss. Technol., 1995, 28, 25 Search PubMed.
- N. J. Miller and C. A. Rice-Evans, Food Chem., 1997, 60, 331 CrossRef CAS.
- T. P. Whitehead, D. Robinson, S. Allaway, J. Syms and A. Hale, Clin. Chem., 1995, 41, 32 Search PubMed.
- S. Ashida, S. Okazaki, W. Tsuzuki and T. Suzuki, Anal. Sci., 1991, 7, 93 Search PubMed.
- A. E. Holley and K. H. Cheeseman, Br. Med. Bull., 1993, 49, 494 Search PubMed.
- B. L. Milic, S. M. Djilas and J. M. Canadanovic-Brunet, Food Chem., 1998, 61, 443 CrossRef CAS.
- M. K. Sharma and G. R. Buettner, Free Rad. Biol. Med., 1993, 14, 649 CrossRef CAS.
- J. M. Coxon and E. Patsalides, Aust. J. Chem., 1982, 35, 509 CAS.
- M. G. Simic, in Autoxidation in Food and Biological Systems, ed. M. S. Simic and M. Karel, Plenum Press, New York, 1980, pp.17–25 Search PubMed.
- M. L. Vicente, J. A. Empis, N. Deighton, S. M. Glidewell, B. A. Goodman and C. C. Rowlands, J. Chem. Soc., Perkin Trans. 2, 1998, 449 RSC.
- G. M. Rosen, E. J. Rauckman and E. Finkelstein, in Autoxidation in Food and Biological Systems, ed. M. S. Simic and M. Karel, Plenum Press, New York, 1980, pp. 71–87 Search PubMed.
- K. M. Schaich and D. C. Borg, in Autoxidation in Food and Biological Systems, ed. M. S. Simic and M. Karel, Plenum Press, New York, 1980, pp 43–70 Search PubMed.
- Y. Noda, K. Anzai, A. Mori, M. Kohno, M. Shinmei and L. Packer, Biochem. Mol. Biol. Int., 1997, 42, 35 Search PubMed.
- P. T. Gardner, D. B. McPhail and G. G. Duthie, J. Sci. Food Agric., 1998, 76, 257 CrossRef CAS.
- P. T. Gardner, D. B. McPhail, A. Crozier and G. G. Duthie, J. Sci. Food Agric., 1999, 79, 1011 CrossRef CAS.
- M. Krishna and A. Samuni, Methods Enzymol., 1994, 234, 580 Search PubMed.
- J. Vasquez-Vivar, J. Joseph, H. Karoui, H. Zhang, J. Miller and P. Martasek, Analusis, 2000, 28, 487 Search PubMed.
- S. Saeed, S. A. Fawthrop and K. H. Nazlin, J. Sci. Food Agric., 1999, 79, 1809 CrossRef CAS.
- G. R. Buettner and B. A. Jurkiewicz, in Handbook of Antioxidants, ed. E. Cadenas and L. Packer, Marcel Dekker, New York, 1996, pp. 91–115 Search PubMed.
- D. M. Lee, Methods Enzymol., 1994, 234, 513 Search PubMed.
- C. A. Rice-Evans and N. J. Miller, Methods Enzymol., 1994, 234, 279 Search PubMed.
- N. J. Miller, in Natural Antioxidants and Food Quality in Atherosclerosis and Cancer Prevention, ed. J. T. Kumpulainen and J. T. Salonen, Royal Society of Chemistry, Cambridge, 1996, pp. 69–72 Search PubMed.
- N. J. Miller and C. A. Rice-Evans, Free Rad. Res., 1997, 26, 195 Search PubMed.
- C. A. Rice-Evans, N. J. Miller, P. G. Bolwell, P. M. Bramley and J. B. Pridham, Free Rad. Res., 1995, 22, 375 Search PubMed.
- R. L. Rouseff, S. F. Martin and C. D. Youtsey, J. Agric. Food Chem., 1987, 35, 1027 CrossRef CAS.
- P. Pietta, P. Simonetti, C. Gardana and P. Mauri, J. Pharm. Biomed. Anal., 2000, 23, 223 CrossRef CAS.
- F. Bonina, C. Puglia, A. Tomaino, A. Saija, N. Mulinacci, A. Romani and F. F. Vincieri, J. Pharm. Pharmacol., 2000, 52, 1279 Search PubMed.
- U. Krings and R. G. Berger, Food Chem., 2001, 72, 223 CrossRef CAS.
- C. Sanchez-Moreno, J. A. Larrauri and F. Saura-Calixto, J. Sci. Food Agric., 1998, 76, 270 CrossRef CAS.
- M. Wettasinghe and F. Shahidi, Food Chem., 2000, 70, 17 CrossRef CAS.
- H. S. Choi, H. S. Song, H. Ukeda and M. Sawamura, J. Agric. Food Chem., 2000, 48, 4156 CrossRef CAS.
- I. I. Koleva, H. A. G. Niederlander and T. A. van Beek, Anal. Chem., 2000, 72, 2323 CrossRef CAS.
- M. N. Peyrat-Maillard, S. Bonnely and C. Berset, Talanta, 2000, 51, 709 CrossRef CAS.
- Y. Sawai and J. H. Moon, J. Agric. Food Chem., 2000, 48, 6247 CrossRef CAS.
- R. Amarowicz, M. Naczk and F. Shahidi, J. Am. Oil Chem. Soc., 2000, 77, 957 Search PubMed.
- C. Da Porto, S. Calligaris, E. Celotti and M. C. Nicoli, J. Agric. Food Chem., 2000, 48, 4241 CrossRef CAS.
- M. W. Lee, Y. A. Lee, H. M. Park, S. H. Toh, E. J. Lee, H. D. Jang and Y. H. Kim, Arch. Pharmacol. Res., 2000, 23, 455 Search PubMed.
- M. I. Gil, F. A. Tomas-Barberan, B. Hess-Pierce, D. M. Holcroft and A. A. Kader, J. Agric. Food Chem., 2000, 48, 4581 CrossRef CAS.
- Y. R. Lu and L. Y. Foo, Food Chem., 2000, 68, 81 CrossRef CAS.
- V. Weber, P. Coudert, C. Rubat, E. Duroux, F. Leal and J. Couquelet, J. Pharm. Pharmacol., 2000, 52, 523 Search PubMed.
- O. Dangles, G. Fargeix and C. Dufour, J. Chem. Soc., Perkin Trans. 2, 2000, 1653 RSC.
- M. F. Wang, Y. Jin and C. T. Ho, J. Agric. Food Chem., 1999, 47, 3974 CrossRef CAS.
- I. F. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239, 70 CrossRef CAS.
- I. F. F. Benzie and J. J. Strain, Methods Enzymol., 1999, 299, 15 Search PubMed.
- P. T. Gardner, T. A. C. White, D. B. McPhail and G. G. Duthie, Food Chem., 2000, 68, 471 CrossRef CAS.
- R. J. De Lange and A. N. Glazer, Anal. Biochem., 1989, 177, 300 CrossRef CAS.
- H. Wang, G. Cao and R. L. Prior, J. Agric. Food Chem., 1996, 44, 701 CrossRef CAS.
- G. Cao, C. P. Verdon, A. H. B. Wu, H. Wang and R. L. Prior, Clin. Chem., 1995, 41, 1738 Search PubMed.
- C. J. Guo, G. H. Cao, E. Sofic and R. L. Prior, J. Agric. Food Chem., 1997, 45, 1787 CrossRef CAS.
- C. R. Caldwell, Anal. Biochem., 2001, 293, 232 CrossRef CAS.
- D. D. M. Wayner, G. W. Burton, K. U. Ingold and S. Locke, FEBS Lett., 1985, 187, 33 CrossRef CAS.
- R. Luukkainen, R. Aejmelaeus, H. Alho, T. Metsa-Ketela, S. R. Ikonen and M. K. Salo, Free Rad. Res., 1999, 30, 189 Search PubMed.
- A. Ghiselli, M. Serafini, G. Maiani, E. Azzini and A. Ferro-Luzzi, Free Rad. Biol. Med., 1995, 18, 29 CrossRef CAS.
- M. Serafini, G. Maiani and A. Ferroluzzi, J. Nutr., 1998, 128, 1003 Search PubMed.
- V. Bohm, Ernaehr.-Umsch., 2000, 47, 372 Search PubMed; Chem. Abstr., 2000, 133, 334216 Search PubMed.
- K. Malminiemi, A. Palomaki and O. Malminiemi, Free Rad. Res., 2000, 33, 581 Search PubMed.
- M. Serafini, J. A. N. Laranjinha, L. M. Almeida and G. Maiani, J. Nutr. Biochem., 2000, 11, 585 CrossRef CAS.
- R. van den Berg, G. R. M. M. Haenen, H. van den Berg and A. Bast, Food Chem., 1999, 66, 511 CrossRef CAS.
- C. Sanchez-Moreno, M. T. Satué-Gracia and E. N. Frankel, J. Agric. Food Chem., 2000, 48, 5581 CrossRef CAS.
- S. P. Boyle, V. L. Dobson, S. J. Duthie, D. C. Hinselwood, J. A. M. Kyle and A. R. Collins, Eur. J. Clin. Nutr., 2000, 54, 774 CrossRef CAS.
- S. R. J. Maxwell, Basic Res. Cardiol., 2000, 95, 65 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |