Recent developments in detection methods for microfabricated analytical devices
Maria A.
Schwarz
and
Peter C.
Hauser
*
The University of Basel, Department of Chemistry, Spitalstrasse 51, 4056, Basel, Switzerland
First published on 9th August 2001
Abstract
Sensitive detection in microfluidic analytical devices is a challenge because of the extremely small detection volumes available. Considerable efforts have been made lately to further address this aspect and to investigate techniques other than fluorescence. Among the newly introduced techniques are the optical methods of chemiluminescence, refraction and thermooptics, as well as the electrochemical methods of amperometry, conductimetry and potentiometry. Developments are also in progress to create miniaturized plasma-emission spectrometers and sensitive detectors for gas-chromatographic separations.
 Peter C. Hauser Peter C. Hauser | Peter C. Hauser obtained a BEng in Chemistry from the Winterthur Engineering College in Switzerland in 1981. This was followed by an MSc in Chemistry at the University of British Columbia in Canada and a PhD from LaTrobe University in Melbourne, Australia. Subsequently he was a postdoctoral fellow in the group of Professor Simon at the ETH in Zurich and then in the Electrochemistry Group at the Paul Scherrer Institute in Switzerland. In 1991 he was appointed as a lecturer at the Chemistry Department of the University of Auckland, New Zealand, and in 1996 to the present position of Associate Professor at the Chemistry Department of the University of Basel. His research interests are in chemical sensors, electrophoretic separation and in the development of miniature and rugged analytical instrumentation for field and process analysis and finding novel ways of applying electronics to analytical instrumentation. |
Introduction
Microscale analytical devices employing a short and narrow micromachined channel for electrophoretic separations (often termed ‘μTAS’ or simply ‘chips’) were introduced in the early 1990s (see, for example, refs. 1–5). In comparison with conventional capillary electrophoresis, much shorter analysis times were reported for these novel devices. Fluorescence detection was employed, presumably because its inherent high sensitivity is a good match for the small detection volumes available. This mode of detection has since been widely used and has been implemented in the early commercial instruments available. Most analyte species do not fluoresce intrinsically, however, and therefore have to be chemically derivatized. The success of fluorescence detection is partly due to its high sensitivity but also to the facts that the analysis of biochemically relevant species is of high interest and that amino acids and related species can be derivatized readily with a fluorescent tag (e.g., with fluorescein isothiocyanate). DNA may be labelled easily with intercalators. For other classes of analytes, however, the technique is not as easily implemented and direct fluorescence detection cannot be considered a universal approach. It is therefore not surprising that in order to extend the use of microfluidic separation devices beyond the now established biochemical applications, much attention is given to the development of other detection techniques. Another driving force behind the further exploration of detection methods for chip-based methods is the goal of overall miniaturization. Fluorescence detection is also not ideal in this regard and not surprisingly a lot of work is currently focusing on electrochemical methods as these represent the most direct approach to the signal conversion.Please note that the scope of this review is limited to developments over approximately the last two years regarding general on-chip detection methods. Other, without doubt also interesting, developments, such as those involving the coupling of microfluidic devices to mass spectrometers, the integration of selective sensors or sensor arrays into microfluidic devices, or coulometric microtitrators, are not covered here. Recent reviews concerning the coupling with mass spectrometers6 and on general developments in microfabricated analytical devices7,8 are available.
Spectroscopic detection methods
Fluorescence
The established way of carrying out fluorescence detection is based on an argon-ion laser with an emission line at 488 nm, a microscope lens for collecting the fluoresced light and a photomultiplier tube for detection. Many authors have indeed continued to use laser induced fluorescence (LIF) in the development of new applications not discussed here. Harrison and co-workers have optimized the optics of this arrangement in order to minimize background intensity and noise and achieved detection of fluorescein at a concentration of 1 pM with their set-up.9 The same group has then adapted this system for use with a red (635 nm) laser diode as excitation source in order to produce a more compact overall system.10 Laser diodes are much smaller than argon lasers and therefore better suited in terms of miniaturization and portability. However, semiconductor lasers are readily available only for the infrared and red regions of the spectrum, which are of limited use for fluorescence measurements. A novel violet laser diode has recently come on the market and its use for detection in conventional capillary electrophoresis has been demonstrated, but at present the limited lifetime and high cost preclude routine applications of this device.11 A further simplification of fluorescence detection can be obtained, presumably at reduced sensitivity, if a blue light-emitting diode (LED) and a silicon photodiode are used instead of laser source and photomultiplier tube, respectively. Webster et al. have recently reported such a combination with detector and optical interference filter constructed on the separation device itself with standard photolithographic techniques in silicon.12 This arrangement has the advantage of the elimination of external optics and alignment procedures but necessitates more elaborate manufacturing.Other authors have worked on the further improvement of the detection limit of laser induced fluorescence detection by implementing photon counting. Single molecule detection was first reported in 1997; however, this was achieved on a large DNA-fragment with multiple labelled sites.13 Fister et al. have demonstrated the detection of individual small fluorescent molecules (Rhodamine B and 6G) separated by electrophoresis.14 Gösch et al. have carried out studies on flow patterns in small channels and have also achieved single molecule detection of a fluorescent dye.15 In a different development, Zugel et al. have demonstrated the possibility of two-photon excited fluorescence detection on chip, a method which requires a fairly elaborate instrumentation but which is also claimed to lead to improved detection limits.16
Postcolumn labelling carried out on-chip with the aid of an auxiliary reagent channel eliminates the manual analyte derivatization step prior to injection. Liu et al. have recently optimized this method for protein determination by use of a dye which binds non-covalently and possesses the fast reaction kinetics necessary for rapid analysis and high sensitivity.17 Lu et al.18 have extended fluorescence detection on chip to inorganic species by determining uranium using a selective calixarene ligand which has been specifically derivatized with a rhodamine moiety in order to render it fluorescent.
In order to make fluorescence detection more universal, a number of researchers have investigated the use of indirect fluorescence detection. For indirect detection a fluorescent substance with a charge of the same sign as that of the analyte is added to the background buffer and its displacement (in order to maintain electroneutrality) is measured in the form of negative going peaks. Munro et al. have demonstrated the determination of amino acids by this means, eliminating the need for derivatization, albeit at lower sensitivity and hence higher detection limits than achieved with direct fluorescence.19 Arundell et al.20 have demonstrated the detection of environmentally relevant phenols. Wallenborg and Bailey21 achieved the sensitive indirect detection of the explosive trinitrotoluene (TNT) and related compounds using a semiconductor laser at 750 nm. The latter application is illustrated in Fig. 1. Sirichai and de Mello22,23 have carried out the analysis of photographic developer solutions via indirect fluorescence detection on chip. While indirect UV-absorption methods are accepted in capillary electrophoresis for the determination of inorganic ions, these should generally be considered as a last resort, because small changes in a background signal are monitored and the methods are thus limited by the noise on the baseline. Higher demands are posed on the stability of the light source in indirect detection than in direct detection and when using shorter pathlengths. It should be borne in mind that indirect laser induced fluorescence has not become accepted for conventional capillary electrophoresis because of shortcomings in the stability of the intensity of available lasers. Nevertheless, indirect fluorescence detection has the potential to become a useful general on-chip detection technique and the situation is likely to improve with the development of new short wavelength semiconductor lasers which are more stable than other types of sources.
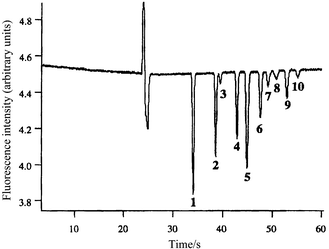 | ||
| Fig. 1 Fast separation and detection of trinitrotoluene (TNT) and related compounds (a mixture of nitrated and aminated toluenes) at 20 ppm using indirect laser induced fluorescence. Reprinted with permission from ref. 21. Copyright 2000 American Chemical Society. | ||
Absorbance
Detection via absorbance is not commonly used for on-chip detection because the short optical pathlengths allow only limited sensitivity. Nevertheless Haswell and co-workers have used this method successfully in their adaptation of classical flow-injection analysis (FIA) methods for the determination of phosphate,24 nitrite25 and nitrate26 to the electroosmotically pumped chip-format. Lu and Collins have detected transition metals electrophoretically separated as their complexes with 4-(2-pyridylazo)resorcinol (PAR) down to about 1 ppm with a green light-emitting diode (LED) as radiation source.27 Two groups have independently developed absorbance detectors based on linear photodiode arrays in order to image the entire separation channel.28,29 This allows the direct visualization of the dynamics of isoelectric focusing28 or separation29 processes.One possible approach to increasing the sensitivity of absorbance measurements is the use of multireflection cells to increase the optical pathlength. Based on an earlier design by Verpoorte et al.,30 Harrison and co-workers31 have recently constructed such a cell in glass with an effective pathlength that is enhanced by a factor of 5–10. A different possibility for achieving higher sensitivity in absorbance measurements is the use of thermooptical methods such as the thermal lens approach. In this technique, requiring a somewhat more elaborate system, the refractive index change of the cell volume on absorption of light (via its heating) is measured by the reflection of a probe beam. The application of this method in conventional capillary electrophoresis has recently been reviewed.32 Kitamori and co-workers have employed thermal lens detection for micromachined channel systems33–36 and have achieved detection limits of approximately 0.1 μM for model dyes.33
Chemiluminescence and other methods
Several groups have investigated chemiluminescence techniques. Greenway et al.37 and Xu et al.38 have presented micro-machined flow-through devices for the detection of codeine and chromium, respectively. These systems rely on the selectivity of the reaction rather than electrophoretic separation. Good detection limits of approximately 10−7M were achieved in both cases and the latter application is illustrated in Fig. 2. Hashimoto et al.39 and Tsukagoshi et al.40 have used the same technique for the detection of electrophoretic separations of amino acids and transition metals, respectively. The method has the advantage of relative instrumental simplicity, as a light source is not required. The use of electrochemiluminescence should be beneficial as even better detection limits may be achieved via rapid electrochemical recycling of reagents.41 Bornhop and co-workers have described a refractive index detector employing changes in the interference pattern created by interaction of a laser beam with the separation channel and demonstrated the detection of some model substances (including carbohydrates) down to about 1 mM.42,43 Manz and co-workers have described an optical imaging technique, involving Fourier transformation, which allows the determination of the velocity of solutes or particles moving in a microchannel.44,45 Trumbull et al. have investigated the use of nuclear magnetic resonance (NMR) as a detection method for a microfabricated electrophoretic device and admit poor detection limits.46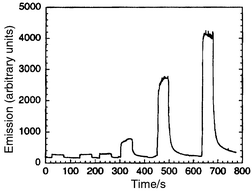 | ||
| Fig. 2 Chemiluminescence signal obtained on a micromachined flow-through device for injections of 1 × 10−7, 1 × 10−6, 5 × 10−6, 1 × 10−5, 5 × 10−5 and 1 × 10−4 M of CrIII. Reprinted with permission from ref. 38. | ||
Electrochemical detection methods
For all electrochemical detection methods the chemical signal is directly converted into the electronic domain via simple electrodes, therefore the overall system can be more compact and the goal of miniaturization can be driven further. On the other hand, practically all commercial instruments for conventional capillary electrophoresis are based on either UV absorption or fluorescence. Overall, miniaturization is not a goal for benchtop instruments and electrochemical detection methods have often been considered incompatible because the combination of high voltages applied for electrophoretic separation and sensitive electrodes is seen as a conflict. However, as will be seen below, it has been found that with appropriate designs of the detector cell the separation voltage does not interfere with the electrochemical measurement. These detection methods have therefore recently found wider acceptance for conventional capillary electrophoresis. Much of the know-how gained can be transferred to the chip platform.Amperometry
Amperometric detection is based on measuring the oxidation or reduction currents of analytes at a working electrode. It has the advantage of generally good detection limits but is restricted to electroactive species. It is the most widely reported electrochemical method for conventional capillary electrophoresis and a number of research groups have recently reported its implementation for microchannel based separations.One possible approach to eliminating any interference by the applied electrical field on the detection is to use a so-called decoupler in front of the detector. These consist of a porous material through which the solution is in contact with the electrophoretic ground electrode. Rossier et al.47 and Chen et al.48 have implemented this approach on micromachined electrophoresis chips with amperometric detection and examined their performance using model substances. On the other hand, most workers using amperometric detection in conventional capillary electrophoresis are now relying on a simpler scheme to avoid electrical interference (for a recent review see ref. 49). It was found that effective suppression of the separation voltage can be achieved without significant band broadening by employing a cell geometry which shows a drastic enlargement of the channel cross-section immediately in front of the detector electrode. The leads to a low resistance in the solution volume between working and electrophoretic ground electrodes and therefore to a small residual voltage at the detector electrode. This approach has been chosen by the majority of the research groups who have transferred amperometric detection to micromachined separation devices and this work is discussed below.
Woolley et al. have demonstrated the detection of neurotransmitters and DNA-fragments on Pt-electrodes patterned on a glass chip for electrophoresis.50 Liu et al. have detected dopamine and hydroquinone on a gold electrode in a study on the effect of dynamic wall coating for an electrophoretic microchip.51 Lunte and coworkers have reported several devices incorporating amperometric detector electrodes and have demonstrated the separation and detection of different classes of biochemically relevant species.52–55 They have developed a method to incorporate carbon electrodes into micromachined separation devices53,54 and carried out dual electrode detection.54,55 Carbon as an electrode material is particularly useful because of its wide anodic potential window but cannot be processed with the usual microfabrication techniques. Dual electrode detection gives some information on the analyte identity when the electrodes are poised at different potentials.
Instead of integrating the detector electrode directly into the electrophoretic separation device (as for the references cited above) Wang and co-workers56-64 and Hilmi and Luong65,66 have used a different arrangement. The separation channel is extended to the edge of the separation device so that an external detector cell can be aligned in a right angle with the distal end. This approach leads to a more complex system and requires alignment by the user, but has the advantages that the electrode preparation does not have to be compatible with the micromachining of the channel system and that the detector electrode can be replaced without having to discard the entire separation system. It is likely that, in general, the lifetimes of the detector electrodes are shorter than those of the separation devices as a whole and therefore that a replaceable external detector cell may be preferable. This is not relevant, of course, if the entire device can be mass-fabricated cheaply. Wang and co-workers have demonstrated a range of applications, which include biochemically and environmentally relevant species as well as chemical warfare agents and explosives.56,57,59,61–64 Hilmi and Luong have also demonstrated the latter application and have shown the feasibility of detecting trinitrotoluene (TNT) and related compounds.65,66
Wang and co-workers have presented a device which incorporates pre-column derivatization in order to render amino acids detectable at a gold working electrode.60 The same authors have also recently extended the fixed potential amperometric detection to the voltammetric mode. This method not only yields information on the redox potential of the analytes but may lead to increased sensitivity through the use of electrolytic preconcentration methods such as anodic stripping voltammetry.58
Schwarz et al. have recently shown that it is possible to use a simplified arrangement based on a total of two electrodes only, the amperometric detector electrode and the electrophoretic ground.67 The latter acts as a counter to the detector and as a pseudo-reference. In view of miniaturization and simplification, the elimination of a conventional reference is particularly important because these require an internal electrolyte solution and liquid junction. The same authors have demonstrated the utility of the copper electrode for the direct on-chip determination of amino acids and carbohydrates,67 species which cannot be determined readily on electrodes based on other materials, as well as the application of amperometric detection for rapid on-chip chiral separations of certain biochemically relevant substances.68 The performance of the two-electrode arrangement for these methods is illustrated in Fig. 3.
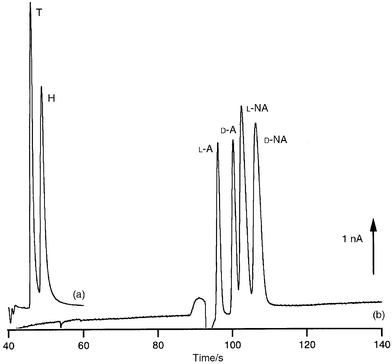 | ||
| Fig. 3 Two electropherograms illustrating amperometric on-chip detection. (a) Direct detection of the amino acids tryptophan (T) and histidin (H) at a copper electrode. (b) Rapid chiral separation of adrenaline (A) and noradrenaline (NA) employing a cyclodextrin with detection at a gold electrode. | ||
Conductimetry
Conductimetric detection has also been investigated for chip-based separation and is a good technique in particular for small (inorganic) ions which are otherwise not readily detected. For conventional capillary electrophoresis conductivity detection has recently seen a renaissance.49,69 Grass and co-workers70,71 have applied micromachining techniques in the construction of devices in poly(methylmethacrylate) (PMMA) for isotachophoresis with conductivity detection. These devices can be regarded as miniaturized versions of channel based systems for isotachophoresis which were constructed with conventional machining techniques well before the introduction of conventional capillary electrophoresis.72 The performance of the new micromachined devices appears to be very similar to those earlier instruments. Prest et al.73,74 have described a similar system for isotachophoresis, in which the detection was carried out by the so-called potential gradient technique. This may be considered an indirect method for conductivity measurement. Isotachophoresis has never been widely accepted, however, perhaps because of relatively high detection limits, and it remains to be seen if it will become more successful in a miniaturized format. Guijt et al. have presented a chip for conventional zone electrophoresis with integrated conductivity detector and demonstrated detection of organic acids down to the low μM-range.75 This method is illustrated in Fig. 4.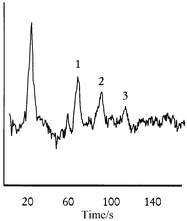 | ||
| Fig. 4 Conductimetric detection of fumaric (1), malic (2) and citric (3) acid (all at 10 μM) on a microfabricated electrophoresis device. Reprinted with permission from ref. 75. | ||
Potentiometry
A potentiometric detector based on a miniaturized ion-selective electrode in a micromachined flow-channel has been demonstrated by Manz and co-workers for the detection of Ba2+ and proposed for detection in electrophoresis chips.76 Special membranes have to be used for this purpose in order to achieve detection of a range of ions rather than the usually desired selectivity to just a single ion. This kind of detector has not been widely used in standard capillary electrophoresis. However, they are a simple alternative, attractive for ions which are not readily detected otherwise, show low conductivity and are not amenable to optical detection.49Detection in the gas phase
Broekaert and co-workers have developed a new microwave-induced plasma (MIP) source which is based on a micromachined fused-silica substrate.77–79 Microwave-induced plasmas are used for atomic emission spectroscopy and are mainly employed as element specific detectors in gas chromatography. The device reported by Broekaert is significantly smaller than the Beenakker cavity usually employed and the power consumption, at 15 W, is lower by a factor of about 10. The application to the detection of mercury (by the cold vapour technique)79 and chloroform vapour (via emission from chlorine)78 has been demonstrated. Note that the device, in contrast to the inductively coupled plasma spectrometers widely used for atomic emission spectrometry, is not tolerant to the introduction of aerosols. Blades and co-workers have demonstrated the feasibility of a capacitively coupled plasma sustained by 10 W of radiofrequency power at 13 MHz in a quartz microchannel.80 Niemax and co-workers have reported a similar device with an even lower power consumption (<1 W), operated at a frequency between 5 and 20 kHz, which was demonstrated for the detection of halogenated hydrocarbons by diode laser absorption spectrometry.81 Eijkel et al. have reported a direct current helium plasma (dc plasma) implemented on a glass chip.82–84 The power consumption of this device is only approximately 10 mW and the emission from diatomic molecules such as CH and CO was used for the detection of methane and also larger hydrocarbons separated by (off-chip) gas chromatography. This application is illustrated in Fig. 5. Gas chromatography, incidentally, represents the earliest analytical technique implemented on a chip format85 and commercial portable instruments incorporating micromachined systems are available. However, the thermal conductivity detection presently employed does not allow trace analysis. In a different approach to address this shortcoming, Yu and Koo have combined a wafer based chromatograph with a novel high sensitivity glow-discharge detector with potentiometric signal read out.86 The detection limits for small hydrocarbons were reported to be in the low ppb range.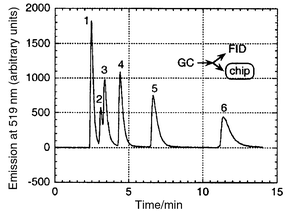 | ||
| Fig. 5 Hexane (1), methanol (2), ethanol (3), 1-propanol (4), 1-butanol (5) and 1-pentanol (6) separated by off-chip chromatography and detected with a micromachined dc-plasma emission spectrometer. Reprinted with permission from ref. 83. Copyright 2000 American Chemical Society. | ||
Conclusions
As has been seen, a multitude of new detection methods for analytical on-chip devices have been reported recently. However, much of the work must be considered preliminary. In many cases the detectors have only been tested for very few model substances and often only marginal electropherograms were presented. Frequently, mass detection limits are reported prominently, as they look very impressive given the very low detection volumes, while the analytically more relevant concentration data is often sobering. As the field is very much in flux, only a snapshot of the current situation can be given and the authors apologize for any oversights.In conventional capillary electrophoresis, the most commonly employed detection method is UV absorption, even though this is often hampered by limited sensitivity because of the short optical pathlengths. The second most commonly used on capillary electrophoresis is laser induced fluorescence (LIF). On-chip, the straightforward absorption measurement is not likely to succeed. In order to ensure wide application of the miniaturized electrophoretic devices, it appears that one or more other simple and robust standard method(s), besides fluorescence, must yet become established. There is, perhaps, also a stronger urge to implement electrochemical methods on microfabricated devices than for standard capillary electrophoresis instruments because the use of electrodes for detection leads to smaller instruments and cost reduction, which is part of the motivation for miniaturization. Microfabrication is well suited for the construction of electrochemical detectors. These are difficult to manufacture by traditional methods, even for conventional capillaries, because of the small dimensions involved, and the better mechanical precision achieved with micromachining should lead to improved analytical performance. Amperometry, conductimetry and, perhaps, electrochemiluminescence are deemed likely to become strong complementary methods to direct and indirect fluorescence detection for chip-based electrophoretic separations.
Acknowledgements
Partial funding for this work was provided by the Swiss National Science Foundation under grant number 2100-055403.98.References
- D. J. Harrison, A. Manz, Z. Fan, H. Lüdi and H. M. Widmer, Anal. Chem., 1992, 64, 1926 CrossRef.
- D. J. Harrison, K. Fluri, K. Seiler, Z. Fan, C. S. Effenhauser and A. Manz, Science (Washington, DC), 1993, 261, 895 Search PubMed.
- K. Seiler, D. J. Harrison and A. Manz, Anal. Chem., 1993, 65, 1481 CrossRef CAS.
- C. S. Effenhauser, A. Manz and H. M. Widmer, Anal. Chem., 1993, 65, 2637 CrossRef CAS.
- S. C. Jacobson, A. W. Moore and J. M. Ramsey, Anal. Chem., 1995, 67, 2059 CrossRef CAS.
- R. D. Oleschuk and D. J. Harrison, Trends Anal. Chem., 2000, 19, 379 CrossRef CAS.
- J. P. Kutter, Trends Anal. Chem., 2000, 19, 352 CrossRef CAS.
- G. J. M. Bruin, Electrophoresis, 2000, 21, 3931 CrossRef CAS.
- G. Ocvirk, T. Tang and D. J. Harrison, Analyst, 1998, 123, 1429 RSC.
- G. Jiang, S. Attiya, G. Ocvirk, W. E. Lee and D. J. Harrison, Biosens. Bioelectron., 2000, 14, 861 CrossRef CAS.
- J. E. Melanson and C. A. Lucy, Analyst, 2000, 125, 1049 RSC.
- J. R. Webster, M. A. Burns, D. T. Burke and C. H. Mastrangelo, Anal. Chem., 2001, 73, 1622 CrossRef CAS.
- C. S. Effenhauser, G. J. M. Bruin, A. Paulus and M. Ehrat, Anal. Chem., 1997, 69, 3451 CrossRef CAS.
- J. C. Fister, S. C. Jacobson, L. M. Davis and J. M. Ramsey, Anal. Chem., 1998, 70, 431 CrossRef CAS.
- M. Gösch, H. Blom, J. Holm, T. Heino and R. Rigler, Anal. Chem., 2000, 72, 3260 CrossRef.
- S. A. Zugel, B. J. Buyrke, F. E. Regnier and F. E. Lytle, Anal. Chem., 2000, 72, 5731 CrossRef CAS.
- Y. Liu, R. S. Foote, S. C. Jacobson, R. S. Ramsey and J. M. Ramsey, Anal. Chem., 2000, 72, 4608 CrossRef CAS.
- Q. Lu, J. H. Callahan and G. E. Collins, Chem. Commun., 2000, 1913 RSC.
- N. J. Munro, Z. Huang, D. N. Finegold and J. P. Landers, Anal. Chem., 2000, 72, 2765 CrossRef CAS.
- M. Arundell, P. D. Whalley and A. Manz, Fresenius' J. Anal. Chem., 2000, 367, 686 CrossRef CAS.
- S. R. Wallenborg and C. G. Bailey, Anal. Chem., 2000, 72, 1872 CrossRef CAS.
- S. Sirichai and A. J. de Mello, Analyst, 2000, 125, 133 RSC.
- S. Sirichai and A. J. de Mello, Electrophoresis, 2001, 22, 348 CrossRef CAS.
- G. N. Doku and S. J. Haswell, Anal. Chim. Acta, 1999, 382, 1 CrossRef CAS.
- G. M. Greenway, S. J. Haswell and P. H. Petsul, Anal. Chim. Acta, 1999, 387, 1 CrossRef CAS.
- P. H. Petsul, G. M. Greenway and S. J. Haswell, Anal. Chim. Acta, 2001, 428, 155 CrossRef CAS.
- Q. Lu and G. E. Collins, Analyst, 2001, 126, 429 RSC.
- Q. Mao and J. Pawliszyn, Analyst, 1999, 124, 637 RSC.
- H. Nakanishi, T. Nishimoto, A. Arai, H. Abe, M. Kanai, Y. Fujiyama and T. Yoshida, Electrophoresis, 2001, 22, 230 CrossRef CAS.
- E. Verpoorte, A. Manz, H. Lüdi, A. E. Bruno, F. Maystre, B. Krattiger, H. M. Widmer, B. H. van der Schoot and N. F. de Rooij, Sens. Actuators, B, 1992, 6, 66 CrossRef.
- H. Salimi-Moosavi, Y. Jiang, L. Lester, G. McKinnon and D. J. Harrison, Electrophoresis, 2000, 21, 1291 CrossRef CAS.
- A. K. Malik and W. Faubel, Chem. Soc. Rev., 2000, 29, 275 RSC.
- K. Sato, H. Kawanishi, M. Tokeshi, T. Kitamori and T. Sawada, Anal. Sci., 1999, 15, 525 CAS.
- H. M. Sorouraddin, A. Hibara, M. A. Proskurnin and T. Kitamori, Anal. Sci., 2000, 16, 1033 CAS.
- M. Tokeshi, T. Minagawa and T. Kitamori, Anal. Chem., 2000, 72, 1711 CrossRef CAS.
- M. Tokeshi, M. Uchida, A. Hibara, A. Sawada and T. Kitamori, Anal. Chem., 2001, 73, 2112 CrossRef CAS.
- G. M. Greenway, L. J. Nelstrop and S. N. Port, Anal. Chim. Acta, 2000, 405, 43 CrossRef CAS.
- Y. Xu, F. G. Bessoth, J. C. T. Eijkel and A. Manz, Analyst, 2000, 125, 677 RSC.
- M. Hashimoto, K. Tsukagoshi, R. Nakajima, K. Kondo and A. Arai, J. Chromatogr. A, 2000, 867, 271 CrossRef CAS.
- K. Tsukagoshi, M. Hashimoto, R. Nakajima and A. Arai, Anal. Sci., 2000, 16, 1111 CAS.
- A. Arora, A. J. de Mello and A. Manz, Anal. Commun., 1997, 34, 393 RSC.
- K. Swinney, D. Markow and D. J. Bornhp, Anal. Chem., 2000, 72, 2690 CrossRef CAS.
- K. Swinney and D. J. Bornhop, Analyst, 2000, 125, 1713 RSC.
- H. J. Crabtree, M. U. Kopp and A. Manz, Anal. Chem., 1999, 71, 2130 CrossRef CAS.
- Y. C. Kwok, N. T. Jefferey and A. Manz, Anal. Chem., 2001, 73, 1748 CrossRef CAS.
- J. D. Trumbull, I. K. Glasgow, D. J. Beebe and R. L. Magin, IEEE Trans. Biomed. Eng., 2000, 47, 3 CrossRef CAS.
- J. S. Rossier, R. Ferrigno and H. H. Girault, J. Electroanal. Chem., 2000, 492, 15 CrossRef CAS.
- D. C. Chen, F. L. Hsu, D. Z. Zhan and C. H. Chen, Anal. Chem., 2001, 73, 758 CrossRef CAS.
- T. Kappes and P. C. Hauser, Electroanalysis, 2000, 12, 165 CrossRef CAS.
- A. T. Woolley, K. Lao, A. N. Glazer and R. A. Mathies, Anal. Chem., 1998, 70, 684 CrossRef CAS.
- Y. Liu, J. C. Fanguy, J. M. Bledsoe and C. S. Henry, Anal. Chem., 2000, 72, 5939 CrossRef CAS.
- C. S. Henry, M. Zhong, S. M. Lunte, M. Kim, H. Bau and J. J. Santiago, Anal. Commun., 1999, 36, 305 RSC.
- R. S. Martin, A. J. Gawron, B. A. Fogarty, F. B. Regan, E. Dempsey and S. M. Lunte, Analyst, 2001, 126, 277 RSC.
- A. J. Gawron, R. S. Martin and S. M. Lunte, Electrophoresis, 2001, 22, 242 CrossRef CAS.
- R. S. Martin, A. J. Gawron, S. M. Lunte and C. S. Henry, Anal. Chem., 2000, 72, 3196 CrossRef CAS.
- J. Wang, B. Tian and E. Sahlin, Anal. Chem., 1999, 71, 5436 CrossRef CAS.
- J. Wang, B. Tian and E. Sahlin, Anal. Chem., 1999, 71, 3901 CrossRef CAS.
- J. Wang, R. Polsky, B. Tian and M. P. Chatrathi, Anal. Chem., 2000, 72, 5285 CrossRef CAS.
- J. Wang, M. P. Chatrathi, B. Tian and R. Polsky, Anal. Chem., 2000, 72, 2514 CrossRef CAS.
- J. Wang, M. P. Chatrathi and B. Tian, Anal. Chem., 2000, 72, 5774 CrossRef CAS.
- J. Wang, M. P. Chatrathi and B. Tian, Anal. Chim. Acta, 2000, 416, 9 CrossRef CAS.
- J. Wang, M. P. Chatrathi, B. Tian and R. Polsky, Electroanalysis, 2000, 12, 691 CrossRef CAS.
- J. Wang, M. P. Chatrathi and B. Tian, Anal. Chem., 2001, 73, 1296 CrossRef CAS.
- J. Wang, M. P. Chatrathi, A. Mulchandani and W. Chen, Anal. Chem., 2001, 73, 1804 CrossRef CAS.
- A. Hilmi and J. H. T. Luong, Environ. Sci. Technol., 2000, 34, 3046 CrossRef CAS.
- A. Hilmi and J. H. T. Luong, Anal. Chem., 2000, 72, 4677 CrossRef CAS.
- M. A. Schwarz, B. Galliker, K. Fluri, T. Kappes and P. C. Hauser, Analyst, 2001, 126, 147 RSC.
- M. Schwarz and P. C. Hauser, J. Chromatogr. B, 2001, submitted for publication.
- A. J. Zemann, Trends Anal. Chem., 2001, 20, 346 CrossRef CAS.
- B. Grass, A. Neyer, M. Jöhnck, D. Siepe, F. Eisenbeiss, G. Weber and R. Hergenröder, Sens. Actuators, B, 2001, 72, 249 CrossRef.
- D. Kaniansky, M. Masar, J. Bielcikova, F. Ivanyi, F. Eisenbeiss, B. Stanislawski, B. Grass, A. Neyer and M. Jöhnck, Anal. Chem., 2000, 3596 CrossRef CAS.
- P. Bocek, M. Deml and J. Janak, J. Chromatogr., 1975, 106, 283 CrossRef CAS.
- J. E. Prest, S. J. Baldock, N. Bektas, P. R. Fielden and B. J. Treves Brown, J. Chromatogr. A, 1999, 836, 59 CrossRef CAS.
- J. E. Prest, S. J. Baldock, P. R. Fielden and B. J. Treves Brown, Analyst, 2001, 126, 433 RSC.
- R. M. Guijt, E. Baltussen, G. van der Steen, R. B. M. Schasfoort, S. Schlautmann, H. A. H. Billiet, J. Frank, G. W. K. van Dedem and A. van den Berg, Electrophoresis, 2001, 22, 235 CrossRef CAS.
- R. Tantra and A. Manz, Anal. Chem., 2000, 72, 2875 CrossRef CAS.
- A. M. Bilgic, U. Engel, E. Voges, M. Kückelheim and J. A. C. Broekaert, Plasma Sources Sci. Technol., 2000, 9, 1 CrossRef CAS.
- A. M. Bilgic, E. Voges, U. Engel and J. A. C. Broekaert, J. Anal. At. Spectrom., 2000, 15, 579 RSC.
- U. Engel, A. M. Bilgic, E. Voges and J. A. C. Broekaert, Anal. Chem., 2000, 72, 193 CrossRef CAS.
- A. Bass, C. Chevalier and M. W. Blades, J. Anal. At. Spectrom., submitted for publication. Search PubMed.
- M. Miclea, K. Kunze, G. Musa, J. Franzke and K. Niemax, Spectrochim. Acta, Part B, 2001, 56, 37 CrossRef.
- J. C. T. Eijkel, H. Stoeri and A. Manz, Anal. Chem., 1999, 71, 2600 CrossRef CAS.
- J. C. T. Eijkel, H. Stoeri and A. Manz, Anal. Chem., 2000, 72, 2547 CrossRef CAS.
- J. C. T. Eijkel, H. Stoeri and A. Manz, J. Anal. At. Spectrom., 2000, 15, 297 RSC.
- S. C. Terry, J. H. Jerman and J. B. Angell, IEEE Trans. Electron Devices, 1979, ED-26, 1880 CrossRef.
- C. M. Yu and J. C. Koo, Presentation at the 9th OnSite Conference, Florida, 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |