DOI:
10.1039/B003189P
(Paper)
J. Mater. Chem., 2001,
11, 160-167
Crystal structure and electronic properties
of Ca4Mn2TiO9.93, an n = 3
Ruddlesden–Popper compound†‡
Received 22nd May 2000, Accepted 29th June 2000
First published on 10th October 2000
Abstract
Traditional solid state synthetic methods were used to prepare
a polycrystalline sample of the n = 3 Ruddlesden–Popper
phase Ca4Mn2TiO9.93. The crystal structure (space
group Pbca, a = 5.31411(5), b = 5.31148(5), c = 26.9138(2) Å)
was determined by the simultaneous analysis of neutron and X-ray diffraction
data, with near-edge anomalous scattering being used to provide contrast
between Mn and Ti cations. The latter show a small preference for the octahedral
sites at the centre of the three-layer perovskite blocks within the structure.
Neutron diffraction data collected at 5 K show no evidence for long-range
magnetic ordering, although an enhanced magnetisation with a weak remanence
is observed at low temperature; this is ascribed to the presence of antisymmetric
exchange interactions. Ca4Mn2TiO9.93 is a
semiconductor with a temperature-dependent activation energy of ∼100 meV.
Only weak (ρB/ρ0 > 0.9
in 14 T at 75 K) magnetoresistance was observed.
Introduction
The magnetotransport properties of Mn-containing oxides have stimulated
a great deal of research activity in recent years.1,2
The majority of the compounds of interest show a spontaneous magnetisation
at low temperatures and contain mixed-valence Mn3+/Mn4+
cations in a perovskite-related crystal structure. It has been demonstrated
that, in many cases, their resistivity at temperatures slightly above the
Curie temperature (Tc) drops by several orders
of magnitude on the application of a magnetic field. No adequate explanation
of this negative colossal magnetoresistance (CMR; (ρB − ρ0)/ρ0 ∼ −0.99 or ρB/ρ0 ∼ 0.01)
behaviour is presently available, although it is generally agreed that double
exchange3 on the Mn sublattice is an important
factor. However, models which rely on this mechanism cannot explain the observation4 of CMR in the pyrochlore Tl2Mn2O7,
which, as an oxide of Mn4+, does not have a non-integral
number of electrons per Mn, as is required if the double exchange mechanism
is to operate. The range of perovskite-related compounds which show the
effect is not limited to those with a formula of the type ABO3.
It also includes5,6 compounds having the
formula A2A′B2O7, that is the n = 2
members of the Ruddlesden–Popper (RP) family7
(A,A′)n + 1BnO3n + 1, where
A and A′ are both large, electropositive cations. The perovskite ABO3
is the n = ∞ end member of this family,
all the members of which can be considered to consist of blocks of corner-sharing
BO6 octahedra which extend to infinity in the xy plane,
and are n octahedra thick parallel to z; neighbouring blocks
are separated by rock-salt layers, so that the formula can usefully be
written as ((A,A′)BO3)n(A,A′)O.
We have previously reported8 the crystal structure
and magnetic properties9 of an n = 3
compound Ca4Mn3O10. This material shows an
orthorhombic distortion of the tetragonal structure usually associated with
RP phases, and it orders as a weak ferromagnet at 115 K. However, despite
the presence of a spontaneous magnetisation, magnetotransport measurements10 revealed a level of magnetoresistance in this compound
which, although significant (ρB/ρ0 ∼ 0.6
in 14 T at 61 K), was not as high as that seen in n = 2
phases. We have subsequently11 prepared the
compounds Ca4Mn2FeO10 − δ
and Sr4Mn2FeO10 − δ
in order to ascertain whether the structural and electronic changes induced
by the introduction of Fe can have a beneficial effect on the magnetotransport
properties of the n = 3 phases; the introduction
of Fe4+ into an oxide of Mn4+ was intended
to lead to the presence of both d4 and d3 cations, as
is the case in the Mn3+/Mn4+ oxides which
show CMR. However, our aims were frustrated by the effects of both Mn/Fe
and anion disorder, together with incomplete cation oxidation. We have now
prepared and characterised a phase, Ca4Mn2TiO10 − δ,
in which 33% of the paramagnetic cations are replaced by diamagnetic
Ti4+. Our hope was that the Mn and Ti cations would order
in such a way that the octahedral sites in the central layer of a perovskite
block were occupied by Ti, with the sites in the two outer layers occupied
by Mn. Conventional single-layer n = 1 RP
phases have previously12 shown charge ordering
and spin-glass behaviour rather than CMR, but we hoped that this new material
containing single, magnetically-isolated layers of MnO6 octahedra
might act as a CMR material with appropriate doping. The synthesis and characterisation
of this phase are described below. The structural part of this study is non-routine
because neither conventional powder X-ray diffraction nor neutron diffraction
can reliably distinguish between Ti and Mn; in both cases the scattering factors
of the two elements are essentially the same. In order to introduce the necessary
contrast into the diffraction experiment we have taken advantage of the tunability
of synchrotron radiation to select an X-ray wavelength close to the absorption
edge of Mn4+
(but not Ti4+). The
form factor of Mn4+ close to the edge can be written as f = f0 + Δf′ + if″, where f0
is the form factor away from the edge, and Δf′ and f″
are the real and imaginary anomalous contributions to the form factor close
to the edge. The latter can be treated as a constant, whereas the former is
a fast function of wavelength and can thus be used to introduce significant
contrast between the scattering by two elements which have a similar number
of electrons.13 Our strategy was therefore
to perform a structure refinement by simultaneously analysing neutron diffraction
data and near-edge X-ray data, with the former providing vital information
about the location of the light anions and the latter defining the distribution
of the Ti and Mn cations; a set of X-ray data collected away from the
edge was also included in the analysis.Experimental
A polycrystalline sample (∼5 g) of Ca4Mn2TiO10 − δ
was prepared using standard ceramic techniques. Stoichiometric quantities
of CaCO3
(99.995%, Alfa), MnO2
(99.999%
Puratronic, Alfa) and TiO2
(99.995% Puratronic,
Alfa) were ground together thoroughly, pelletized, loaded into an alumina
crucible and heated in air at temperatures of up to 1275![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C as
described in Table 1. After the
final firing the sample was cooled to 1200°C at a rate of 5°C min−1
and then air quenched to room temperature. The progress of the reaction was
monitored using an in-house X-ray powder diffractometer. We were unable
to determine the oxygen content of the sample reliably by chemical methods.
°C as
described in Table 1. After the
final firing the sample was cooled to 1200°C at a rate of 5°C min−1
and then air quenched to room temperature. The progress of the reaction was
monitored using an in-house X-ray powder diffractometer. We were unable
to determine the oxygen content of the sample reliably by chemical methods.
Table 1 Synthesis conditions for Ca4Mn2TiO10 − δ
| Temperature/°C | Firing time/h |
|---|
| 800 | 36 |
| 1000 | 24 |
| 1200 | 60 |
| 1250 | 40 |
| 1275 | 84 |
The final product was subjected to a more extensive X-ray examination
using diffractometer 2.3 at the Daresbury Laboratory Synchrotron Radiation
Source. Data were collected by stepping (Δ2θ = 0.01°)
over the angular range 20 ≤ 2θ/° ≤ 120
at nominal wavelengths of λ = 1.4000 and
1.8930 Å, the latter value being chosen to lie close to the Mn
K absorption edge. Data were also collected on SrMnO3, a well-characterised14 oxide known to contain six-coordinate Mn4+.
The data collected on this standard facilitated calibration of the anomalous
contribution (Δf′) to the form factor of Mn4+
close to the absorption edge. The background fluorescence signal from the
sample was compared with that from the standard in order to confirm that the
absorption edges are essentially coincident in the two materials. Neutron
powder diffraction data were collected on Ca4Mn2TiO10 − δ
at 295 K and 5 K using the diffractometer D2b at the ILL, Grenoble.
The sample was contained in a cylindrical vanadium can of diameter 8 mm
and data were collected with λ = 1.5940 Å,
8 ≤ 2θ/° ≤ 150, Δ2θ = 0.05°.
Magnetisation data were collected on Ca4Mn2TiO10 − δ
as a function of applied field and temperature using a Quantum Design MPMS
SQUID magnetometer. M(T) was measured in a field
of 100 G whilst warming through the temperature range 5 ≤ T/K ≤ 300;
data were collected after cooling in zero field (ZFC) and after
cooling in the measuring field (FC). M(H)
was measured (−20 ≤ H/kG ≤ 20)
at temperatures of 300, 100, 30, 10 and 5 K (in that order)
with field cooling between measurements; these temperatures were selected
after examination of the M(T) data. Magnetotransport
data were collected on a sintered bar of dimensions 5 × 2 × 2 mm
using apparatus described elsewhere.15 Standard
four probe geometry was used, with the direct current (≤50 µA)
perpendicular to the applied field. ρ(T) was
measured while cooling the material from room temperature to 4.2 K
in zero field and ρB was then measured sequentially
at temperatures of 75, 100, 150 and 200 ± 1 K over
the field range 0 ≤ B/T ≤ 14; warming
between temperatures was performed in zero field.
Results
Structural chemistry
The analysis of the powder diffraction data was carried out using the GSAS
program package.16 In all cases the background
level was modelled using a shifted Chebyshev polynomial, and the peak shapes
were described by a pseudo-Voigt function. The neutron diffraction data
collected at room temperature were used in a preliminary structure refinement.
This gave no reliable information on the Mn/Ti distribution, but allowed
us to deduce accurate unit cell parameters and structural parameters for the
oxygen and calcium atoms. These parameters were then used in the analysis
of the X-ray data collected at λ = 1.4000 Å,
the Mn/Ti distribution being assumed to be random. This strategy demonstrated
that the nominal values of the neutron and X-ray wavelengths were self-consistent
to four decimal places. Consideration of the peak positions in the X-ray
data set collected on Ca4Mn2TiO10 − δ
close to the absorption edge established the higher wavelength as 1.8930 Å,
again in excellent agreement with the intended value. Having calibrated both
of the X-ray wavelengths, the structure of SrMnO3 was refined
making simultaneous use of the two available data sets (λ = 1.4000
and 1.8930 Å), the purpose being to determine a value for Δf′,
the real part of the anomalous scattering close to the absorption edge. The
imaginary contribution to the Mn4+ form factor at λ = 1.8930 Å
(f″)
was calculated (using GSAS) to be 2.81 e− per Mn4+
and was held constant at this value in the data analysis. Refinement of the
usual profile parameters, atomic coordinates, isotropic temperature factors
and (at the higher wavelength only)
Δf′ resulted
in the fitting parameters Rwpr = 6.00%
(λ = 1.4000 Å),
7.03%
(λ = 1.8930 Å),
6.49%
(combined) and χ2 = 1.328,
with Δf′ taking a value of −9.04(5) e−
per Mn4+. The high-quality fits obtained for the two data
sets demonstrate that the use of these values for Δf′
and f″ adequately accounts for the change in the form factor
of Mn4+ when a wavelength close to the edge is selected; a
similar value for Δf′ has been determined previously
for Mn4+.17The room temperature structure of Ca4Mn2TiO10 − δ
was then analysed in detail by means of a simultaneous refinement of three
diffraction profiles, that is one neutron and two X-ray data sets. During
these refinements, Δf′ was held constant at the value
determined during the refinement of SrMnO3. The preliminary analysis
of the neutron dataset had shown that, like Ca4Mn3O10,
Ca4Mn2TiO10 − δ
is modelled best using the orthorhombic space group Pbca. However,
there were significant differences between the observed and calculated profiles
when a single-phase model was used. Careful inspection of the data suggested
the presence of a second phase related to the perovskite CaMnO3 − δ.18,19 Refinement of the concentration and unit
cell parameters of an additional phase, formulated as CaMn2/3Ti1/3O2.97
and with the atomic coordinates held constant at the values reported for CaMnO2.97,
resulted in a significant improvement in the fit; the concentration refined
to a value of 1.13% by weight and the unit cell parameters to a = 5.316(1); b = 5.308(1); c = 7.528(1) Å in space group Pbnm.
The overall stoichiometry of the reaction mixture dictates that if an n = ∞
RP phase is present, then there should be also a trace of RP material with n < 3,
but this was not detectable. The simultaneous use of three data sets in our
final refinements made it possible to refine the fractional occupancies of
Mn4+ and Ti4+ over the six-coordinate
4b and 8c sites. There is a small excess of Ti on the 4b sites at the centres
of the perovskite blocks, with a corresponding excess of Mn on the 8c sites
which make up the outer layers of the blocks. Refinement of the oxygen site
occupancies revealed incomplete occupation of the O(9) position;
this is an equatorial anion site in the outer layer of the perovskite blocks.
The composition of the majority phase deduced from these refinements is thus
Ca4Mn2TiO9.93. It follows that 0.14 moles
of Mn3+ are present per mole of Ca4Mn2TiO9.93
and this, together with the presence of the CaMn2/3Ti1/3O2.98
impurity, introduces a small, unquantifiable error into the anomalous dispersion
analysis. The majority of the atoms were modelled with isotropic thermal parameters,
although an anisotropic model was used when trial refinements showed it to
be necessary. The resultant (total) residual fit parameters in the
three-histogram refinement were as follows: Rwp = 6.62%, Rp = 5.64%,
DWd = 1.308 and χ2red = 1.25.
The refined values of the atomic coordinates are listed in Table 2
and the thermal parameters are available as ESI.‡
The corresponding bond lengths and bond angles are listed in Table 3.
A polyhedral view of the structure is shown in Fig. 1
and the observed and calculated diffraction profiles are drawn in Fig. 2.
 |
| | Fig. 1 Polyhedral representation
of the crystal structure of the orthorhombic n = 3
Ruddlesden–Popper phase Ca4Mn3O10. | |
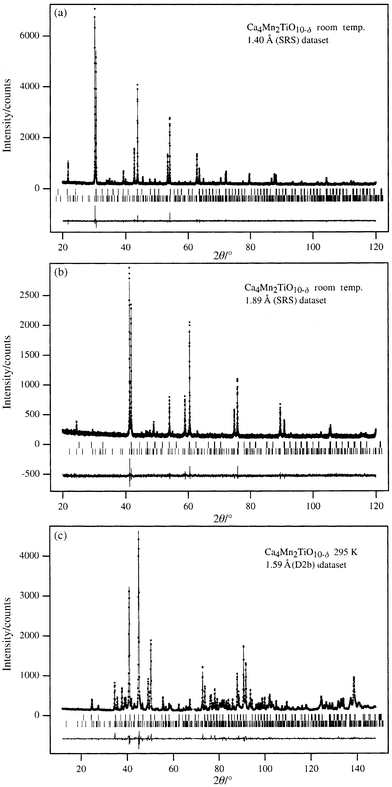 |
| | Fig. 2 Observed, calculated
and difference diffraction profiles for Ca4Mn2TiO9.93
at 295 K: (a) X-rays, λ = 1.4000 Å, (b)
X-rays, λ = 1.8930 Å, (c)
neutrons. Reflection positions for the minority (upper) and majority (lower)
phases are marked. | |
Table 2 Structural parameters for Ca4Mn2TiO9.93
at room temperaturea
| Atom | Site | Occupancy (%) | x | y | z | Uiso/Ueq/Å2 |
|---|
| a = 5.31411(5); b = 5.31148(5); c = 26.9138(2) Å; V = 759.662(16) Å3. |
|---|
| Ca(1) | 8c | 100 | 0.5011(5) | 0.4792(6) | 0.07000(9) | 0.0132 |
| Ca(2) | 8c | 100 | −0.0036(6) | 0.0130(8) | 0.20432(6) | 0.0121 |
| Mn(3) | 4b | 59.8(2.6) | 0 | 1/2 | 0 | 0.0034(6) |
| Ti(4) | 4b | 40.2(2.6) | 0 | 1/2 | 0 | 0.0034(6) |
| Mn(5) | 8c | 70.1(1.3) | 0.4992(7) | 0.0011(7) | 0.14111(7) | 0.0064(4) |
| Ti(6) | 8c | 29.9(1.3) | 0.4992(7) | 0.0011(7) | 0.14111(7) | 0.0064(4) |
| O(7) | 8c | 100 | 0.4425(3) | 0.0017(5) | 0.06987(9) | 0.0036(4) |
| O(8) | 8c | 100 | 0.5432(5) | −0.0124(6) | 0.21117(6) | 0.0063(5) |
| O(9) | 8c | 96.3(8) | 0.7809(7) | 0.2210(6) | 0.13265(9) | 0.0143 |
| O(10) | 8c | 100 | 0.2178(5) | 0.7790(6) | 0.14743(8) | 0.0145 |
| O(11) | 8c | 100 | 0.2874(6) | 0.2909(6) | −0.00860(8) | 0.0063(5) |
Table 3 Selected bond lengths (Å)
and angles (°) for Ca4Mn2TiO9.93
at 295 Ka
| Ca(1)–O(7)i | 2.555(5) | Ca(2)–O(8)i | 2.419(4) | Mn(3)b–O(7) | 1.905(2)
×2 |
| Ca(1)–O(7)ii | 2.793(5) | Ca(2)–O(8)ii | 2.915(4) | Mn(3)–O(11) | 1.903(5)
×2 |
| Ca(1)–O(7)iii | 2.361(3) | Ca(2)–O(8)iii | 2.805(6) | Mn(3)–O(11) | 1.928(5)
×2 |
| Ca(1)–O(7)iv | 2.959(3) | Ca(2)–O(8)iv | 2.536(6) | Mn(5)–O(7) | 1.941(3) |
| Ca(1)–O(9)i | 2.633(4) | Ca(2)–O(8)v | 2.292(2) | Mn(5)–O(8) | 1.901(3) |
| Ca(1)–O(9)ii | 2.415(4) | Ca(2)–O(9)i | 2.501(4) | Mn(5)–O(9)c | 1.912(5) |
| Ca(1)–O(10)i | 3.024(4) | Ca(2)–O(9)ii | 2.880(4) | Mn(5)–O(9) | 1.905(6) |
| Ca(1)–O(10)ii | 2.613(4) | Ca(2)–O(10)i | 2.297(4) | Mn(5)–O(10)c | 1.912(6) |
| Ca(1)–O(11)i | 2.601(4) | Ca(2)–O(10)ii | 2.578(5) | Mn(5)–O(10) | 1.881(5) |
| Ca(1)–O(11)ii | 3.093(4) | | | | |
| Ca(1)–O(11)iii | 2.341(4) | | | | |
| Ca(1)–O(11)iv | 2.666(4) | | | | |
| Superscripts relate to
the oxygen atoms numbered as in Fig. 8(a)
and (b). Mn(3)
is used as shorthand for Mn(3)/Ti(4) with fractional
occupancies given in Table 2. One of the two trans O(9), O(10)
pairs. |
|---|
| O(7)–Mn(3)–O(7) | 180 | O(8)–Mn(5)–O(9) | 92.6(2) |
| O(7)–Mn(3)–O(11) | 89.7(1) | O(8)–Mn(5)–O(9) | 90.8(2) |
| O(7)–Mn(3)–O(11) | 88.8(1) | O(8)–Mn(5)–O(10) | 89.1(2) |
| O(11)–Mn(3)–O(11) | 89.02(4) | O(8)–Mn(5)–O(10) | 90.9(2) |
| | | O(9)–Mn(5)–O(9) | 89.0(2) |
| O(7)–Mn(5)–O(8) | 177.2(3) | O(9)–Mn(5)–O(10) | 178.2(2) |
| O(7)–Mn(5)–O(9) | 90.2(2) | O(9)–Mn(5)–O(10) | 90.7(2) |
| O(7)–Mn(5)–O(9) | 88.8(2) | O(9)–Mn(5)–O(10) | 90.5(2) |
| O(7)–Mn(5)–O(10) | 88.1(2) | O(9)–Mn(5)–O(10) | 178.3(2) |
| O(7)–Mn(5)–O(10) | 89.6(2) | O(10)–Mn(5)–O(10) | 89.8(2) |
| | | | |
| Mn(3)–O(11)–Mn(3) | 157.4(1) | Mn(5)–O(9)–Mn(5) | 160.5(2) |
| Mn(3)–O(7)–Mn(5) | 161.9(2) | Mn(5)–O(10)–Mn(5) | 162.9(2) |
In the absence of low-temperature X-ray data, the Mn/Ti distribution
established at room temperature was carried through to the analysis of the
neutron diffraction data collected at 5 K. The fitting parameters resulting
from the single-histogram Rietveld refinement of the crystal structure
of Ca4Mn2TiO9.93 at 5 K were as follows: Rwp = 5.63%, Rp = 4.32%, DWd = 0.482
and χ2red = 6.232. No
magnetic Bragg peaks were apparent in the data collected at low temperature.
The refined atomic coordinates are listed in Table 4,
the thermal parameters are available as ESI,‡
and the most important bond lengths are given in Table 5.
The observed and calculated diffraction profiles are drawn in Fig. 3.
 |
| | Fig. 3 Observed, calculated
and difference neutron diffraction profiles for Ca4Mn2TiO9.93
at 5 K. Reflection positions for the minority (upper) and
majority (lower) phases are marked. | |
Table 4 Structural parameters for Ca4Mn2TiO9.93
at 5 K
| Atom | Site | Fraction (%) | x | y | z | Uiso/Ueq/Å2 |
|---|
| a = 5.3050(1); b = 5.3047(1); c = 26.8619(6) Å; V = 755.93(5) Å3. |
|---|
| Ca(1) | 8c | 100 | 0.5015(9) | 0.476(1) | 0.0699(3) | 0.0084 |
| Ca(2) | 8c | 100 | −0.004(1) | 0.015(2) | 0.2041 (2) | 0.0082 |
| Mn(3) | 4b | 59.8(2.6) | 0 | 1/2 | 0 | 0.000(2) |
| Ti(4) | 4b | 40.2(2.6) | 0 | 1/2 | 0 | 0.000(2) |
| Mn(5) | 8c | 70.1(1.3) | 0.503(2) | 0.009(2) | 0.1410(3) | 0.003(1) |
| Ti(6) | 8c | 29.9(1.3) | 0.503(2) | 0.009(2) | 0.1410(3) | 0.003(1) |
| O(7) | 8c | 100 | 0.4403(6) | 0.0019(8) | 0.0701(2) | −0.0005(8) |
| O(8) | 8c | 100 | 0.5455(9) | −0.013(1) | 0.2113(1) | 0.001(1) |
| O(9) | 8c | 96.3(8) | 0.783(1) | 0.222(1) | 0.1322(2) | 0.0085 |
| O(10) | 8c | 100 | 0.2158(9) | 0.778(1) | 0.1476(2) | 0.0101 |
| O(11) | 8c | 100 | 0.285(1) | 0.294(1) | −0.0092(2) | 0.003(1) |
Table 5 Selected bond lengths (Å)
for Ca4Mn2TiO9.93 at 5 Ka
| Superscripts relate to
the oxygen atoms numbered as in Fig. 8(a)
and (b). Mn(3)
is used as shorthand for Mn(3)/Ti(4) with fractional
occupancies given in Table 2. One of the two trans O(9), O(10)
pairs. |
|---|
| Ca(1)–O(7)i | 2.54(1) | Ca(2)–O(8)i | 2.401(7) | Mn(3)b–O(7) | 1.910(5)
×2 |
| Ca(1)–O(7)ii | 2.81(1) | Ca(2)–O(8)ii | 2.926(7) | Mn(3)–O(11) | 1.881(8)
×2 |
| Ca(1)–O(7)iii | 2.348(5) | Ca(2)–O(8)iii | 2.82(1) | Mn(3)–O(11) | 1.948(7)
×2 |
| Ca(1)–O(7)iv | 2.964(5) | Ca(2)–O(8)iv | 2.52(1) | Mn(5)–O(7) | 1.932(8) |
| Ca(1)–O(9)i | 2.619(8) | Ca(2)–O(8)v | 2.292(5) | Mn(5)–O(8) | 1.906(8) |
| Ca(1)–O(9)ii | 2.408(8) | Ca(2)–O(9)i | 2.492(8) | Mn(5)–O(9)c | 1.88(1) |
| Ca(1)–O(10)i | 3.036(8) | Ca(2)–O(9)ii | 2.889(8) | Mn(5)–O(9) | 1.91(1) |
| Ca(1)–O(10)ii | 2.607(8) | Ca(2)–O(10)i | 2.290(8) | Mn(5)–O(10)c | 1.96(1) |
| Ca(1)–O(11)i | 2.605(8) | Ca(2)–O(10)ii | 2.567(8) | Mn(5)–O(10) | 1.85(1) |
| Ca(1)–O(11)ii | 3.108(7) | | | | |
| Ca(1)–O(11)iii | 2.330(8) | | | | |
| Ca(1)–O(11)iv | 2.639(8) | | | | |
Magnetism and magnetotransport
The temperature dependence of the molar magnetic susceptibility, defined
as M/H, of Ca4Mn2TiO9.93
is shown in Fig. 4. Above 245 K
the data can be fitted to a Curie–Weiss Law with Cm = 4.70(6) cm3 K mol−1, θ = −365(9) K;
this value of the Curie constant is greater than the spin-only value (3.91 cm3 K mol−1).
The molar magnetisation is clearly enhanced in the temperature range 5 < T/K < 70.
Comparison of the ZFC and FC signals shows the presence of hysteresis below
13 K, and both show a local maximum at ∼8 K. The field dependence
of the magnetisation at five selected temperatures is shown in Fig. 5.
At 300, 100 and 30 K, M(H) is linear, with
no hysteresis evident. However, at 10 K and 5 K the linearity
is lost, and hysteresis is observed at low fields; M(H)
remains symmetrical about the origin. |
| | Fig. 4 Molar magnetic susceptibility
of Ca4Mn2TiO9.93 as a function of temperature,
measured in a field of 100 G; the inset shows a fit of the inverse
susceptibility to the Curie–Weiss law. Fit parameters are given in the
text. | |
 |
| | Fig. 5 (a) Field
dependence of magnetisation of Ca4Mn2TiO9.93
for −20 ≤ H/kG ≤ 20, at temperatures
of 300, 100, 30, 10 and 5 K. (b) Field dependence of magnetisation
of Ca4Mn2TiO9.93 for –0.5 ≤ H/kG ≤ 0.5,
at temperatures of 10 and 5 K. | |
The magnetotransport behaviour of Ca4Mn2TiO9.93,
as a function of field at four different temperatures, is shown in Fig. 6. The sample resistance was immeasurably
high (∼300 MΩ) below 70 K. It proved possible
to model the temperature dependence of the resistivity of Ca4Mn2TiO9.93
in the temperature regions 125 < T/K < 295
and 77 < T/K < 123 using a simple
activated conduction law ρ ∝ exp(EA/kT)
with EA = 126.75(5) and 96.5(1) meV
respectively.
 |
| | Fig. 6 Field dependence of
normalised resistivity ρ(B)/ρ(0)
of Ca4Mn2TiO9.93 at temperatures of 200,
150, 100 and 75 K. The inset shows the temperature dependence of resistivity
at zero field. | |
Discussion
Partial (33%) substitution of Ti for Mn in Ca4Mn3O10
results in the retention of orthorhombic symmetry, in contrast to Fe substitution
which restores the ideal tetragonal symmetry of the n = 3
RP phase, albeit with the introduction of positional disorder on the anion
sublattice; this disorder is not seen in Ca4Mn2TiO9.93.
Whereas the Fe cations showed a significant preference for the sites at the
centre of the perovskite blocks in Ca4Mn2FeO9.75,
the degree of cation ordering in the Ti-doped material is only significant
at the 2σ level, with the dopant again preferring the central
layer. The octahedra in the central layer of the perovskite blocks of Ca4Mn2TiO9.93
are larger and less regular than those in the undoped sample. The increase
in the mean bond length from 1.899 Å to 1.912 Å
can simply be attributed to the relatively large ionic radius of Ti4+,
but the anisotropic nature of the increase is interesting. In Ca4Ti3O1020 and Ca4Mn3O10 − δ
the Mn–O distances in these centrosymmetric, central octahedra are all
equal (within σ), whereas in Ca4Mn2TiO9.93
although the lengths of the two apical Mn/Ti–O(7) bonds
and two of the equatorial Mn/Ti–O(11) bonds are essentially
the same (∼1.90 Å), the other pair of Mn/Ti–O(11)
distances are significantly longer (∼1.93 Å). The
origin of this distortion is unclear. The mean Mn/Ti–O bond length
in the outer octahedra is 1.909 Å, lower than that in the central
octahedra, and thus consistent with the outer site holding a lower concentration
of the larger Ti4+ cations. The outer octahedra in Ca4Mn2TiO9.93
are less regular than the central octahedra, a pattern which was also observed
in the cases of Ca4Ti3O10 and Ca4Mn3O10 − δ.
However, the nature of the irregularity is different. Whereas in Ca4Ti3O10
and Ca4Mn3O10 − δ
the trans equatorial bonds in the outer layer differ significantly
from each other in length, such that the cation can be considered to be displaced
from the centre of the octahedron, in Ca4Mn2TiO9.93
three of the four equatorial bonds are essentially equal (∼1.91 Å),
with one Mn/Ti–O(10) distance (1.881 Å)
being significantly shorter. As in Ca4Ti3O10
and Ca4Mn3O10 − δ,
the length of the apical Mn/Ti–O(8) bond extending from
the outer octahedron into the rocksalt layer is shorter than the trans
Mn/Ti–O(7) bond directed into the perovskite block. The
magnitude of this asymmetry, the origin of which has been discussed previously,8 increases with increasing Ti content in the three
compounds under discussion; this is consistent with the known bonding properties
of the Ti4+ ∶ 3d0 cation, which
favours asymmetric coordination with multiple bonding to oxygen. Although
there is a considerable spread among the Mn/Ti–O distances, the cis
Mn–O–Mn bond angles are all close to 90°. However, the trans
Mn–O–Mn angles are typically ∼160°, and the relative rotation
of neighbouring octahedra illustrated in Fig. 7
is a significant aspect of the orthorhombic distortion, being comparable in
magnitude to that seen in Ca4Mn3O10. The
refined oxygen vacancy concentration allows the presence of 0.14 moles
of the Mn3+: 3d4 cation per mole of Ca4Mn2TiO9.93.
This Jahn–Teller ion might be expected to occupy preferentially the
outer octahedra of the triple layer, where the inherent lack of symmetry facilitates
a distorted geometry. This would be consistent with the concentration of the
oxide anion vacancies on the O(9) site in this layer and the fact
that the thermal parameters (Table 2)
of the atoms in and around the outer layer of the perovskite blocks (Ca(1),
Ca(2), Mn(5), O(9), O(10)) are
larger than those of the atoms located within the central layer (O(11),
Mn(3)). The tilting of the Mn/Ti polyhedra is largely responsible
for the irregularity of the coordination geometry around the calcium sites
illustrated in Fig. 8. In an ideal n = 3
RP structure, the Ca(1) cation in the perovskite block of Ca4Mn2TiO9.93
would be 12-coordinate, but in Ca4Mn2TiO9.93
only ten anions lie within 3 Å of the Ca(1) site. However,
the Ca(2) site within the rock-salt layer achieves the ideal
coordination number of nine within a radius of 2.915(4) Å.
We have previously argued8,11 that the
orthorhombic distortion of the ideal n = 3 structure
in the case of Ca4Mn3O10 is driven by the
need to satisfy the coordination requirements of Ca2+, and
that the absence of such a distortion in Ca4FeMn2O10
can be attributed to the high concentration of Jahn–Teller active Fe4+:
3d4 cations which produce the necessary distortions locally, without
a cooperative phase change. This is consistent with the observation of orthorhombic
symmetry in Ca4Mn2TiO9.93, which has only
a low concentration of Jahn–Teller cations.![Tilting of Mn/TiO6
octahedra viewed along directions close to (a)
[110] and (b)
[11̄0]](/image/article/2001/JM/b003189p/b003189p-f7.gif) |
| | Fig. 7 Tilting of Mn/TiO6
octahedra viewed along directions close to (a)
[110] and (b)
[1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] 0] | |
 |
| | Fig. 8 Coordination geometry
around the cations (a) Ca(1) and (b) Ca(2)
in Ca4Mn2TiO9.93 at room temperature. | |
The crystal structure of Ca4Mn2TiO9.93
undergoes a number of modifications on cooling to 5 K. The mean Mn/Ti–O
bond distance in the central octahedron does not change significantly, but
the mismatch between the cis bonds to O(11) within the
equatorial plane becomes more pronounced. Neither does the mean Mn/Ti–O
bond length (1.91 Å) in the outer octahedron change,
but the cation is now clearly displaced from the centre of the octahedron
along each of the three axes, thus increasing the structural similarity with
Ca4Ti3O10 and Ca4Mn3O10 − δ.
One of the Mn/Ti–O(10) bond lengths is consequently reduced
to 1.85(1) Å, a short distance which is consistent
with the presence of a relatively high Mn concentration on this site. All
of the thermal parameters are reduced on cooling, but those of Ca(1),
Ca(2), O(9) and O(10) remain large enough to
suggest that local static disorder is present at these sites.
The magnetic properties of Ca4Mn2TiO9.93
differ markedly from those of Ca4Mn2FeO9.75
and Ca4Mn3O10 − δ,
both of which show G-type antiferromagnetic order within the perovskite
block (although for the Fe compound the central layer of manganese-poor
octahedra remained magnetically disordered). In contrast, the absence
of magnetic Bragg scattering in the neutron diffraction pattern collected
at 5 K shows that long range magnetic order is not present in Ca4Mn2TiO9.93.
However, the temperature and field dependences of the magnetisation demonstrate
that the sample is not a simple paramagnet. It is instructive to review the
magnetic exchange interactions present in each of the three compounds in order
to rationalise the differences in magnetic behaviour. The observation of insulating,
rather than metallic, behaviour in each case precludes any significant contribution
from double exchange. Orthorhombic Ca4Mn3O10 − δ
has been9 shown to undergo a transition to
a weakly ferromagnetic state at ∼115 K, the spontaneous magnetisation (2 × 10−3μB per Mn at 5 K in 500 G) developing as a result
of antisymmetric exchange (the Dzyaloshinski–Moriya (DM)
mechanism21,22). However, in this
compound the antiferromagnetic t2g–t2g
π interactions
between Mn4+: 3d3 cations are dominant. In Ca4Mn2FeO9.75,
in addition to the antiferromagnetic t2g–t2g
π
interactions between Mn4+ cations, there will be competing,
ferromagnetic superexchange interactions between a half-occupied eg
orbital on Fe4+
(or the small amount of Fe3+
present) and an empty eg orbital on Mn4+.
However, the tetragonal symmetry of this compound prohibits a ferromagnetic
DM interaction. The Fe4+/Fe3+ distributions
in this compound were shown11 to be influential
in determining which of the competing magnetic interactions predominated.
No ordered moment was observed on the Fe-rich octahedral sites in the
central layer of the perovskite block, suggesting overall frustration of the
interactions between the Mn4+: d3 and Fe4+:
d4 cations. However, with Fen+ occupying
only ∼21% of the octahedra in the outer layer of the perovskite
block, antiferromagnetic t2g–t2g
π interactions
between Mn4+ cations predominate and G-type ordering is
established in these layers. With these observations in mind, the competing
interactions within a Mn4+ cation array which has been diluted
with diamagnetic Ti4+: d0 cations, can be evaluated.
We expect the antiferromagnetic π interactions between the numerous Mn4+
cations to dominate, although some ferromagnetic Mn3+ t2g3
eg1–O2−–Mn4+
t2g3
σ superexchange interactions will also be
present as a result of oxygen non-stoichiometry. The dilution of the dominant
antiferromagnetic interactions may be considered to be exacerbated by the
low degree of Mn/Ti ordering over the two transition metal sites, in that
whereas the preference of the Fe cations for the central layer of octahedra
in Ca4FeMn2O10, and the correspondingly low
Fe concentration in the outer octahedra, permitted a solid solution with ∼78%
Mn occupation to preserve a magnetically ordered backbone on the latter sites,
the reduction of the Mn concentration to ∼60% and ∼70%
respectively in the inner and outer layers of Ca4Mn2TiO9.93
prevents the onset of long-range magnetic order. However, these concentrations
are both well in excess of the percolation limit, and it is likely that other
factors are also at work. These could include the modifications to the superexchange
interactions which will be caused by the structural distortions present in
the Mn/TiO6 network. However, next-nearest-neighbour (NNN)
interactions are likely to be a more significant factor. It has been shown23,24 that 50% dilution of the magnetic
sublattice of a perovskite (n = ∞ RP)
by d0 cations results in the formation of a spin glass with Tg ∼ 20 K.
Given the non-frustrated topology of the perovskite structure, this behaviour
is usually ascribed to competition between nearest neighbour (NN)
interactions and the weaker but more numerous NNN interactions. Furthermore,
it has been shown that the NNN interactions are relatively strong when the
magnetic cation has a d3 electron configuration25
and it is therefore reasonable to postulate that competition between NN and
NNN superexchange interactions over a 33% diluted Mn4+:
3d3 magnetic sublattice in a structure which is intermediate between
2- and 3-dimensional prevents the onset of long-range magnetic
order.
Although magnetic ordering is not observed by powder neutron diffraction
above 5 K, the Curie–Weiss parameters deduced from the high temperature
region of the susceptibility data demonstrate that strong intercation magnetic
interactions are present in orthorhombic Ca4Mn2TiO9.93
throughout the measured temperature range. The large, negative Weiss constant
indicates that these short-range interactions are predominantly antiferromagnetic
above 100 K, but the enhancement of the susceptibility observed below
this temperature indicates that, in some way or other, local regions within
the sample develop an enhanced magnetisation. The data show a transition at
13 K, and the presence of a remanent magnetisation below this temperature.
The symmetrical disposition of the hysteresis loop about the origin suggests
the presence of a weak ferromagnetism rather than a spin glass phase; this
contrasts with the case of tetragonal Ca4FeMn2O10,
where displaced hysteresis loops were observed. The magnitude of the magnetisation
per Mn cation at 5 K and 500 G is comparable to that produced
by antisymmetric exchange in orthorhombic Ca4Mn3O10 − δ.
It has been shown9 that this weak DM magnetisation
persists within the perovskite blocks of Ca4Mn3O10 − δ
above the temperature of the transition to a phase showing long-range,
3D magnetic ordering. We therefore propose that at high temperatures, antiferromagnetic
Mn–O–Mn interactions within the perovskite blocks dominate the
short-range magnetism of Ca4Mn2TiO9.93,
but that below ∼100 K the consequences of the antisymmetric DM
exchange become observable, with the magnetisation vector produced by the
non-linear Mn–O–Mn interactions within each block being aligned
with those of neighbouring blocks by the applied field. We envisage the antiferromagnetically
coupled spin components to lie within the perovskite layers, as they do in
both Ca4Mn3O10 − δ
and Ca4FeMn2O10, and the net magnetisation
to lie perpendicular to the layers, as was assumed (though not proved)
in the case of Ca4Mn3O10 − δ.
In order for this model to be valid, any antiferromagnetic interblock coupling
between the z components of the moment must be weaker than is the
case in Ca4Mn3O10 − δ
in order to allow the alignment of the DM moment by relatively weak fields.
This is consistent with the absence of a long-range antiferromagnetic
backbone at low temperatures. The nature of the transition at 13 K
is still unexplained. It is possible that it corresponds to spontaneous alignment
of the weak ferromagnetic moments, which are certainly too weak to be detected
by neutron diffraction. Alternatively, the irreversibility of magnetic behaviour
may be evidence for the blocking of clusters of spins below a temperature
of ∼13 K. Ca4Mn2TiO9.93 would
benefit from more extensive characterisation by magnetometry and μSR spectroscopy
in order to establish the true nature of the magnetic ground state. It is
striking that the introduction of Ti4+, merely a non-magnetic
diluant, has a more dramatic effect on the magnetic properties of Ca4Mn3O10 − δ
than does Fe4+, a magnetic cation which introduces competing
superexchange interactions. This may stem from the introduction of Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O
bonds, and the consequent change in the electron density distribution within
the crystal. Note that it has been assumed throughout this discussion of the
magnetic behaviour of the sample that there is no contribution from the low-level
CaMn2/3Ti1/3O2.98 impurity.
O
bonds, and the consequent change in the electron density distribution within
the crystal. Note that it has been assumed throughout this discussion of the
magnetic behaviour of the sample that there is no contribution from the low-level
CaMn2/3Ti1/3O2.98 impurity.
The magnetotransport behaviour of the Ti-doped phase shows the insulating
behaviour characteristic of a localised-electron system. The value EA = 126.75 meV
measured in the temperature range 125 ≤ T/K ≤ 295
is more typical of an n = 2, rather than an n = 3,
layered manganite.26 This gives some idea
of the narrowing of the bandwidth which occurs on introducing the Ti dopant
into the Mn–O network. Energy fluctuations caused by the randomness
of the chemical substitution and the concomitant crystallographic distortions
will both play a rôle in carrier localisation, but it is noteworthy
that the activation energy is reduced in the temperature region where the
magnetisation is enhanced. Given that negative magnetoresistance is often
associated with the presence of ferromagnetic coupling, it may be significant
that the magnitude of the effect in Ca4Mn2TiO9.93
is comparable to that observed in Ca4Mn3O10 − δ
above the magnetic ordering temperature (115 K) when each
perovskite block carries a weak DM magnetisation, but the interblock coupling
of the atomic moments is negligible, that is when the situation is directly
comparable to that found in Ca4Mn2TiO9.93
over a wide temperature range. We take this as evidence in support of the
model of the magnetic behaviour proposed above.
Acknowledgements
We are grateful to EPSRC, Oxford University and (AIC)
the ORS Award Scheme for financial support.References
- R. M. Kusters, J. Singleton, D. A. Keen, R. McGreevy and W. Hayes, Physica B, 1989, 155, 363 CrossRef CAS.
- A. P. Ramirez, J. Phys.: Condens. Matter, 1997, 9, 8171 Search PubMed.
- C. Zener, Phys.
Rev., 1951, 82, 403 CrossRef CAS.
- Y. Shimakawa, Y. Kubo and T. Manako, Nature, 1996, 379, 53 CrossRef CAS.
- P. D. Battle, S. J. Blundell, M.
A. Green, W. Hayes, M. Honold, A. K. Klehe, N. S. Laskey, J. E. Millburn, L. Murphy, M. J. Rosseinsky, N.
A. Samarin, J. Singleton, N. A. Sluchanko, S. P. Sullivan and J. F. Vente, J. Phys.: Condens.
Matter, 1996, 8, L427 Search PubMed.
- Y. Moritomo, A. Asamitsu, H. Kuwahara and Y. Tokura, Nature, 1996, 380, 141 CrossRef CAS.
- S.
N. Ruddlesden and P. Popper, Acta Crystallogr., 1958, 11, 541.
- P. D. Battle, M. A. Green, J. Lago, J.
E. Millburn, M. J. Rosseinsky and J. F. Vente, Chem. Mater., 1998, 10, 658 CrossRef CAS.
- J. Lago, P. D. Battle and M.
J. Rosseinsky, J. Phys.: Condens.
Matter, 2000, 12, 2505 Search PubMed.
- A. I. Mihut, L. E. Spring, R. I. Bewley, S. J. Blundell, W. Hayes, T. Jestädt, B. W. Lovett, R. McDonald, F. L. Pratt, J. Singleton, P. D. Battle, J. Lago, M. J. Rosseinsky and J.
F. Vente, J. Phys.: Condens. Matter, 1998, 10, L727 Search PubMed.
- P.
D. Battle, W. R. Branford, A. Mihut, M. J. Rosseinsky, J. Singleton, J. Sloan, L. E. Spring and J. F. Vente, Chem. Mater., 1999, 11, 674 CrossRef CAS.
- J. C. Bouloux, J. L. Soubeyroux, A. Daoudi and G. L. Flem, Mater. Res. Bull., 1981, 16, 855 CrossRef CAS.
- M. A. G. Aranda, D. C. Sinclair, J. P. Attfield and A. P. Mackenzie, Phys. Rev.
B, 1995, 51, 12747 CrossRef.
- P. D. Battle, T.
C. Gibb and C. W. Jones, J. Solid State Chem., 1988, 74, 60 CAS.
- L. E. Spring, D. Phil. Thesis, 1999, Oxford University, Oxford,
UK..
- A.
C. Larson and R. B. von Dreele, General Structure Analysis System (GSAS),
Los Alamos National Laboratories, Report LAUR 86-748, 1990..
- Y. Murakami, H. Kawada, H. Kawata, M. Tanaka, T. Arima, Y. Moritomo and Y. Tokura, Phys.
Rev. Lett., 1998, 80, 1932 CrossRef CAS.
- K. R. Poeppelmeier, M.
E. Leonowicz, J. C. Scanlon, W. B. Yelon and J. M. Longo, J. Solid State Chem., 1982, 45, 71 CrossRef CAS.
- H. Taguchi, J. Solid State Chem., 1996, 124, 360 CrossRef CAS.
- M. M. Elcombe, E. H. Kisi, K.
D. Hawkins, T. J. White, P. Goodman and S. Matheson, Acta Crystallogr., Sect. B, 1991, 47, 305 CrossRef.
- I. Dzyaloshinski, J. Phys. Chem. Solids, 1958, 4, 241 CrossRef.
- T. Moriya, Phys. Rev., 1960, 120, 91 CrossRef.
- P. D. Battle, T. C. Gibb, A. J. Herod, S.-H. Kim and P. H. Munns, J. Mater. Chem., 1995, 5, 865 RSC.
- E. J. Cussen, J. F. Vente, P. D. Battle and T. C. Gibb, J. Mater. Chem., 1997, 7, 459 RSC.
- P. D. Battle, J. B. Goodenough and R. Price, J. Solid State Chem., 1983, 46, 234 CrossRef CAS.
- A.
I. Coldea, L. E. Spring, S. J. Blundell, J. Singleton and W. Hayes, J. Phys.:
Condens. Matter, 1999, 11, 674 Search PubMed.
Footnotes |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29
September, 2000, University of Cambridge, UK. |
| ‡ Electronic supplementary information (ESI) available: anisotropic
thermal parameters for Ca4Mn2TiO9.93 at room
temperature and 5 K. See http://www.rsc.org/suppdata/jm/b0/b003189p/ |
| § Present address: Chemistry Department, Liverpool University, Liverpool,
UK L69 7ZD. |
| ¶ Present address: Departamento de Fisica Aplicada, Cinvestav-IPN
Unidad Mérida, Carretera Ant. a Progreso km 6, Apartado Postal #73
Cordemex, Mérida, Yucatan, 97310 México. |
|
| This journal is © The Royal Society of Chemistry 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.