Self-propagating high temperature synthesis of BaFe12O19, Mg0.5Zn0.5Fe2O4 and Li0.5Fe2.5O4; time resolved X-ray diffraction studies (TRXRD)†
Ivan P. Parkina, Quentin A. Pankhurstb, Louise Afflecka, Marco D. Aguasa and Maxim V. Kuznetsovb
aDepartment of Chemistry, Christopher
Ingold Laboratory, University College London, 20 Gordon Street, London, UK WC1H 0AJ
bDepartment of Physics and Astronomy, University College London, Gower
Street, London, UK WC1E 6BT
First published on UnassignedUnassigned3rd October 2000
Abstract
Barium ferrite (BaFe12O19), lithium ferrite (Li0.5Fe2.5O4) and magnesium zinc ferrite (Mg0.5Zn0.5Fe2O4) have been prepared by self-propagating high-temperature synthesis (SHS) reactions from iron, iron(III) oxide and metal oxides and peroxides. The driving force for the reactions is the oxidation of iron powder. Reactions were carried out under an oxygen flow and with the addition of sodium perchlorate as an internal oxidising agent. Reactions were also carried out in the presence of an applied magnetic field of 1.1 T. Pre-organisation of the reactant mixture in an applied field led to increases in synthesis wave velocity (from 2 to 5 mm s−1) and temperature. The reactions were studied by time resolved X-ray powder diffraction on Station 16.4 of the CLRC Daresbury Laboratory. A white beam of X-rays was used in combination with three fixed energy sensitive detectors. Spectra were acquired with scan intervals of 100–250 ms and an effective 2θ range of 10–60°. Transformation of reactants to products occurred on the order of 200 ms in all of the systems studied with the exception of the applied field barium ferrite synthesis where an intermediate of Fe3O4 was observed. Lattice expansion effects during SHS enabled the rates of cooling in the system to be investigated. All of the materials synthesised by SHS were examined both before and after annealing by X-ray powder diffraction, energy dispersive X-ray analysis (EDXA), scanning electron microscopy (SEM), FTIR, Mössbauer spectroscopy and vibrating sample magnetometry.
Solid state reactions require prolonged heating even at elevated temperatures because diffusion within solids is slow.1 Conventional synthesis, also known as the ‘ceramic method’, involves many grinding, heating and cooling steps, in order to overcome the solid state diffusion barrier. Many synthetic routes have been developed to improve upon the ceramic method, such as chemical co-precipitation2,3 and glass crystallisation.4,5 These techniques aim to reduce the solid state diffusion length by mixing the components on an atomic scale. Self-propagating high-temperature synthesis (SHS) is an alternative solid state route that makes use of the exothermicity of a chemical reaction.6
Self-propagating high-temperature synthesis was investigated in
the late 1960s by Merzhanov and co-workers and has become a separate branch
of scientific study.7 SHS enables the preparation
of a wide variety of materials, such as inter-metallic compounds, metal
carbides, oxides, borides and nitrides.8 The
nature of SHS also allows novel applications, for example, coating the inside
of pipes, or containment of radioactive material.9
SHS reactions are highly exothermic. Enough heat is released in the reaction
to ignite successive layers of reaction mixture; hence no external heating
is required. The reactions are characterised by rapid heating and cooling
and may be ignited in two ways—at a point or in bulk. From point initiation
the reaction proceeds via a propagation (also known as synthesis
and combustion) wave which moves through the reaction mixture at velocities
from a few mm s−1 to tens of cm s−1.
Temperatures in excess of 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C are reached. The products require
minimal treatment to produce single-phase materials. With bulk initiation
the reactants are heated in a furnace; once the ignition temperature is reached
the reaction is initiated in a variety of locations. Often this process is
named ‘thermal flash’.
°C are reached. The products require
minimal treatment to produce single-phase materials. With bulk initiation
the reactants are heated in a furnace; once the ignition temperature is reached
the reaction is initiated in a variety of locations. Often this process is
named ‘thermal flash’.
We are interested in the formation of ferrite materials by SHS, particularly
BaFe12O19, Mg0.5Zn0.5Fe2O4
and Li0.5Fe2.5O4.10–12
Ferrites are magnetic oxides that find many uses in commercial applications.
Hard ferrites such as the hexagonal ferrites have high coercivities, making
them useful as permanent magnets, and in applications where demagnetizing
forces must be resisted. Soft ferrites such as garnets and spinels have very
low coercivities, so they find application in systems where magnetic hysteresis
must be kept to a minimum. M-Type hexagonal barium ferrite, BaFe12O19,
was first studied in an industrial context by the Philips Laboratories in
1952.10 It has the magnetoplumbite structure
and is ferrimagnetic. It is a hard magnetic material with a maximum coercivity
of 4800 Oe13 and finds commercial application
as a permanent magnetic material, on swipe cards and in television sets. Mg0.5Zn0.5Fe2O4
has applications in mobile phones and television set focussing systems due
to its high saturation magnetisation of 59.3 emu g−1
and low coercivity. Li0.5Fe2.5O4 has the
cubic inverse spinel structure, with lithium and three-fifths of the Fe3+
ions occupying octahedral sites. It is a soft ferrite with a very high Curie
temperature of 638°C.14 These
features mean it finds applications in microwave and memory-core devices.
Two crystallographic forms of Li0.5Fe2.5O4
have been identified: a superstructured form in which the lithium and iron
atoms are ordered, and a disordered form in which lithium and iron have a
random statistical distribution over all the octahedral positions.15
We have synthesised several different materials by SHS, including BaFe12O19,10 SrFe12O19,10 Li0.5Fe2.5O412 and Mg0.5Zn0.5Fe2O4.11 We have recently made a series of spinel ferrites.16 We began performing SHS reactions in an applied magnetic field in 1997.10 This is a novel area of work; the only previously published results were for increased temperatures and reaction rates for SHS of titanium carbide17 and strontium ferrite.18 This has led in 2000 to the report of ferrite systems synthesised in large external magnetic fields of 15 T.19
A range of intriguing effects has been observed by the use of an applied magnetic field on SHS reactions. The amount of superstructured Li0.5Fe2.5O4 produced depends on whether it was synthesised in an applied field;14 for CuFe2O4 the ratio of tetragonal to cubic phase was found to increase on applied field synthesis;16 and the inversion parameter for MgFe2O4 is influenced by the applied field.16 The bulk magnetic properties are also affected by the application of a field. It is not certain why the external field has these effects. One explanation is that in an applied field the green mixture adopts an hour glass shape within the field, within this shape the magnetic components are organised along field lines. This may give a particularly easy propagation direction for the SHS reaction compared to zero field reactions and hence a different product microstructure. A further and more intriguing explanation is the fact that the applied field could influence the magnetic fields formed at the reaction front during SHS. SHS reactions have been found to produce small magnetic fields, of the order of a few nT, during the synthesis of both magnetic and non-magnetic materials. These so-called chemomagnetic fields are attributed to the movement of ions and electrons at the reaction front.20
External magnetic fields have also been seen to affect mass transport and fractal morphology effects in the electrochemical deposition of copper.21,22 Reduced yields and changed morphologies of carbon nanotubes and fullerenes were observed with the application of magnetic fields during synthesis by arc discharge.23
Much work has been published on SHS combustion theory by Merzhanov6–8,24 and modelling by Bowen
and Derby25 and Yi and Moore.26
It is possible to predict which reactions will undergo SHS. Novikov24 found the empirical relation that the adiabatic
combustion temperature must exceed 1500°C for a reaction to propagate.
However, there is much to be learnt about specific systems. In particular,
it would be interesting to discover information about the reaction pathways
taken during SHS, determine a timescale for the transformations and estimate
reaction temperatures from thermal expansion effects. Time resolved X-ray
diffraction (TRXRD) is a technique that is able to probe these questions.
TRXRD experiments on SHS reactions have been performed on just a handful of occasions using synchrotron radiation, at Brookhaven on the Ta–C system,27 and at LURE, Orsay on the Mo–Si,28 Fe–Al,29 and Al–Ni–Ti30 systems. A monochromatic X-ray beam and fixed detectors were used, typically with a range of 30° 2θ and ca. 30–1000 ms resolution. Kachelmyer et al.31 performed time resolved experiments using a laboratory Cu tube X-ray source, using a limited 2θ range of ca. 10° and time resolution on the order of 1 s.
In this paper we present the first time resolved X-ray diffraction studies (TRXRD) on the synthesis of three different ferrites by SHS. These are also the first time resolved studies of SHS reactions using a white beam of X-rays, conducted in air, and the first to be studied in an external magnetic field. We also compare the effects of doing the SHS synthesis in zero field and an applied field of 1.1 T.
Experimental
All chemicals were obtained from Aldrich Chemical Co. and used as supplied. Manipulations, weighing and grindings were performed in a Saffron Scientific glove box under a nitrogen atmosphere. Sintering was carried out on ground powders in a Carbolite rapid heating furnace with heating and cooling rates of 20°C min−1. Samples were ground after
the SHS reaction and also after sintering; all measurements were recorded
on powder samples. For the applied field SHS reactions a permanent Halbach
cylinder magnet made by Magnetic Solutions Limited was used. This cylinder,
comprising eight NdFeB magnets, provided a field of 1.1 T transverse
to the cylinder axis, with a homogeneity over a 20 mm bore cross section
of better than ±0.4%. A ceramic tile, containing the green mixture,
was placed inside a quartz tube. The tube was then inserted into the Halbach
cylinder prior to initiation of the combustion process.The powders were analysed by X-ray diffraction in the reflection mode on a Philips X-Pert using unfiltered Cu Kα radiation (λ1 = 1.5405 Å, λ2 = 1.5443 Å). Vibrating sample magnetometry was carried out on an Aerosonic 3001 magnetometer at room temperature in applied fields of up to 7.5 kOe. Scanning electron micrographs and electron microprobe data were obtained on both a Hitachi S-4000 with an energy dispersive analysis attachment and on a JEOL EMA. 57Fe Mössbauer spectra were recorded with a Wissel MR-260 constant acceleration spectrometer with a triangular drive waveform. Spectra were folded to remove baseline curvature and were calibrated relative to α-iron at room temperature. FT-IR spectra were obtained as KBr pellets on a Nicolet 205. Synthesis wave temperatures were determined by optical pyrometry and FLIR thermal imaging camera.
Reactions in zero magnetic field
°C).
A flow of oxygen at ∼2 l min−1 was supplied
over the reaction area. This induced an orange propagation wave (920°C)
which moved across the mixture at ∼2 mm s−1.
The post-SHS products were ground and sintered at 1150°C for
6 hours.°C) wave with velocity
7 mm s−1 was observed. The post-SHS products
were ground and washed with ∼1 l of distilled water to remove the
NaCl. They were then dried and sintered at 1200°C for 2 hours.°C), ca. 1–2 mm s−1,
was observed. The post-SHS products were ground and sintered at 1150°C
for 6 hours.Reactions in an applied magnetic field
The same reactions were also studied in an applied magnetic field using a 1.1 T permanent magnet with a 20 mm diameter cylindrical bore. The magnetic field direction is transverse to the cylinder axis and oriented vertically. Reactions were conducted in the magnet using a quartz tube and an oxygen flow of ∼2 l min−1. The green mixture aligned with the field direction in an hour glass shape. The low angle detector geometry on Station 16.4 enabled these reactions in a magnetic field to be studied.Time resolved X-ray diffraction
The time resolved X-ray diffraction experiments were performed at CLRC Daresbury Laboratory on Station 16.4. A white beam of X-rays was used with a diameter of 0.1 mm. The beam was at 90° to the direction of the SHS propagation wave (for zero field reactions) and parallel to the cylindrical axis of the magnet (for applied field reactions). Three energy sensitive detectors were placed at fixed low angles of 1.8°, 4.7° and 7.5° and sampled a 1 to 4 mm lozenge shape of the target mixture. Diffraction patterns were collected with a time resolution of up to 100 ms. The experiment set-up is shown in Fig. 1.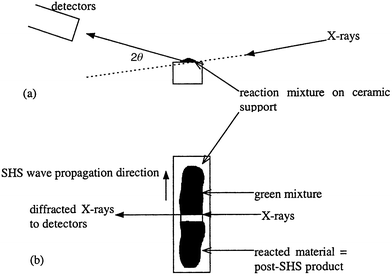 | ||
| Fig. 1 Experimental set-up for the time resolved X-ray powder diffraction studies: (a) X-ray beam and detector set up, (b) propagation wave direction and position of the hot zone for a zero field reaction. | ||
Reactions were conducted on both powders and pressed discs (to 1 ton) of green mixture for each of the three systems.
The X-ray beam was also used to check the homogeneity of the green mixture in the applied field. By positioning the beam at various distances from one end of the quartz tube along the field direction (i.e. vertically), it was possible to check for macroscopic compositional variation within the green mixture.
CAUTION: Care must be taken with NaClO4 as it is a very strong oxidising agent which can react very violently with certain metal powders. SHS reactions are exothermic and material may be ejected up to 1 m from the reaction area. Appropriate precautions should be taken.
Results
Barium ferrite
°C
as measured by optical pyrometry and 920°C by FLIR thermal imaging
camera. The product was a brown powder. Analysis of the powder after the reaction
revealed that it was made up of a mixture of components including BaFe2O4,
FeO and Fe3O4 as well as some unreacted Fe and Fe2O3.
After sintering at 1150°C for 6 h the mixed phase powder
formed single-phase BaFe12O19, eqn. (1).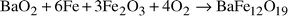 | (1) |
°C as recorded by
optical pyrometry and 1240°C as recorded by a thermal imaging
camera. The product from the reaction was fused and had two distinct looking
parts: a shiny, fused part from the portion in the centre of the magnetic
field, and a matt brown part, Fig. 2.
The fibrous appearance of the shiny product suggests alignment of the initial
powder with the magnetic field lines. The shiny and matt parts were manually
separated and analysed. Large area EDAX scans of both portions showed no overall
element segregation—similar Ba ∶ Fe ratios were observed
in both parts. XRD and Mössbauer analysis revealed that the shiny part
is more fully combusted than the matt part. In both the matt and the shiny
components BaFe2O4, FeO , Fe2O3
and Fe3O4 were detected. In the matt component unreacted
iron metal was also detected, indicating an incomplete combustion reaction.
Electron microprobe analysis revealed that the shiny part has a different
microstructure to the matt applied field and zero field products. It consists
of acicular grains with a Ba ∶ Fe ratio of ∼1 ∶ 12
as required for BaFe12O19, although we conclude from
other analyses that the grains are made up of BaFe2O4
and Fe3O4, which readily combine upon sintering to form
BaFe12O19. VSM measurements have shown that the AF shiny
part, after annealing, has a reduced coercivity compared to the other two
parts, Table 1. | ||
| Fig. 2 Photographs of the fused products from the SHS reaction of BaO2, Fe and Fe2O3 showing the shiny and matt components. | ||
| Lattice parameters/Å | Magnetic parameters | |||||||
|---|---|---|---|---|---|---|---|---|
| Ferrite system | Conditions | XRD of sintered products | a | c | σmax/emu g−1 | σr/emu g−1 | Hc/Oe | Mössbauer |
| BaFe12O19 | Literature | BaFe12O19 | 5.8972(15) | 23.2116(32) | 46 | 35.1 | 2665 | 12k; 4fiv; 4fvi, 2a and 2b sextet subspectra |
| BaO2 + Fe + Fe2O3 | ZF SHS 1150°C/6 h | BaFe12O19 | 5.885(4) | 23.171(4) | 49 | 32 | 2400 | 12k; 4fiv; 4fvi, 2a and 2b sextet subspectra |
| BaO2 + Fe + Fe2O3 | AF SHS 1150°C/6 h | BaFe12O19 | 5.889(4) | 23.190(4) | 45 | 30 | 1800 | 12k; 4fiv; 4fvi, 2a and 2b sextet subspectra |
| Mg0.5Zn0.5Fe2O4 | Literature | Mg0.5Zn0.5Fe2O4 | 8.415(4) | — | 59.3 | Sextet | ||
| MgO + ZnO + Fe2O3 + Fe + NaClO4 | ZF | Mg0.5Zn0.5Fe2O4 | 8.416(4) | — | 59.8 | 1.42 | 9.4 | Sextet |
| MgO + ZnO + Fe2O3 + Fe + NaClO4 | AF | Mg0.5Zn0.5Fe2O4 | 8.415(4) | — | 82.6 | 1.80 | 9.3 | Sextet |
| Li0.5Fe2.5O4 | Literature | Li0.5Fe2.5O4 | 8.331(4) | — | 65 | |||
| Li2O2 + Fe + Fe2O3 | ZF | Li0.5Fe2.5O4 | 8.3121(4) | — | 51.6 | 9.3 | 18.4 | Octahedral and tetrahedral site sextets |
| Li2O2 + Fe + Fe2O3 | AF | Li0.5Fe2.5O4 | 8.3285(4) | — | 54.7 | 5.6 | 14.5 | Octahedral and tetrahedral site sextets |
The SHS reaction did not go to completion in either the applied field or
zero field barium ferrite synthesis. In the applied field reaction the product
was more fully combusted. The solids produced after SHS in both the zero field
and applied field reactions were, however, easily processed into barium ferrite
by heating at 1150°C for 6 h. Both zero field and applied
field samples indexed well (Table 1)
compared to literature values and commercial materials. The Mössbauer
spectra of both the zero field and the applied field SHS sintered barium ferrite
showed a series of overlapping sextets; these sextets could be deconvoluted
into five sub-components with a 12 ∶ 4 ∶ 4 ∶ 2 ∶ 2
ratio corresponding to the 12k; 4fiv; 4fvi, 2a and 2b
sites in hexagonal barium ferrite.10 The coercivity
of BaFe12O19 synthesised in an applied field was significantly
lower than that synthesised in zero field and commercial samples (Table 1).10
A homogeneity check of the green mixture inside the applied field prior to SHS showed identical diffraction patterns measured at various distances from the magnet wall along the field direction, i.e. on a macroscopic scale the applied field green mixture was homogeneous.
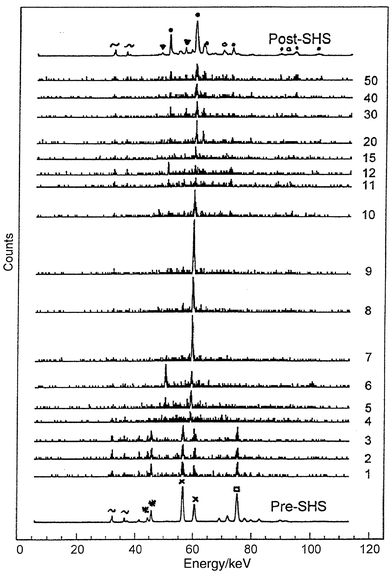 | ||
| Fig. 3 Time resolved X-ray powder diffraction stack plots for the SHS reaction of BaO2, Fe and Fe2O3 obtained at 250 ms intervals. The numbering scheme on the right hand side of the plot shows the run number, the reaction passes through the X-ray beam between runs 3 and 4. The x-axis scale is in energy (keV), the y-axis is intensity (and time). Key: ∼ Ba fluorescence; ▼ BaFe2O4; ● Fe3O4; ○ Fe1 − xO; * BaO2; × Fe2O3; □ Fe. | ||
Fig. 4 is another (and to our
knowledge, unique) way of representing the TRXRD data. Each diffraction
pattern is shown as a series of light and dark lines, the more intense the
peak, the darker the line. This figure clearly shows an abrupt change in diffraction
peaks corresponding to the passage of the synthesis wave through the X-ray
beam. It also displays a curvature to the diffraction peaks towards higher
energies, which corresponds to changes in d-spacings. The d-spacings
are 2.5% larger at the start of the transformation compared to the
post-SHS product. This has been used to estimate the temperatures reached
in the reaction, by taking into account the thermal expansion coefficient
of Fe3O4 that corresponds to those peaks. Based on these
effects a maximum reaction temperature in excess of 2000°C is
calculated. The curvature of the diffraction peaks also gives an estimate
of the cooling rates in these systems. This indicates that the reaction takes
only 60 seconds to cool from well in excess of 2000°C to
100°C. This rapid heating and cooling rate is typical of SHS and
solid state metathesis reactions.6–9
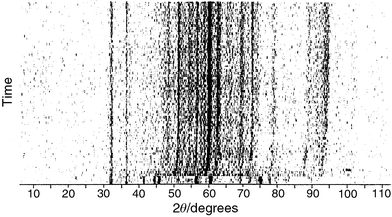 | ||
| Fig. 4 Position of the X-ray diffraction maxima (x-axis) against time (y-axis) for the SHS reaction of BaO2, Fe and Fe2O3. The darker the spot the more intense the diffraction peak. | ||
Magnesium zinc ferrite
°C was measured by
thermal imaging camera. The green mixture in the applied field reactions was
pulled into the magnetic field to adopt an hour glass shape in cross section.
The applied field reactions proceed with a yellow–orange synthesis wave
of velocity ∼10 mm s−1. Again, the reaction
was too fast to observe by optical pyrometry. A temperature of 1280°C
was measured by thermal imaging camera. Sodium perchlorate acts as a wetting
agent and internal source of oxygen and provides extra energy for the reaction.
The reaction failed to propagate in the absence of sodium perchlorate. The
product in both zero field and applied field reactions is brown and slightly
fused. Washing the product with water removes the co-produced sodium chloride
and leaves single-phase magnesium zinc ferrite, eqn. (2).
Notably in these systems the magnesium zinc ferrite is formed directly in
the reaction and did not require further sintering to insure complete reaction.
Essentially single-phase material was obtained directly from the SHS process.
The magnesium zinc ferrite was shown to adopt the cubic spinel stucture with
a cell constant equal to conventionally prepared material (Table 1). EDAX and electron probe measurements
on the triturated powders showed only magnesium, zinc and iron in the correct
molar ratios (oxygen was below the machine cut off). No sodium or
chlorine was detected by these methods. 57Fe Mössbauer was
in agreement with the spinel model and was fitted to iron in octahedral and
tetrahedral sites.11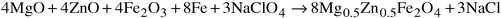 | (2) |
 | ||
| Fig. 5 Photographs of the SHS reaction of MgO, ZnO, Fe, Fe2O3 and NaClO4 in pellet form under a flow of oxygen. The pellet has a diameter of 13 mm and the photographs were obtained at 2 s intervals. | ||
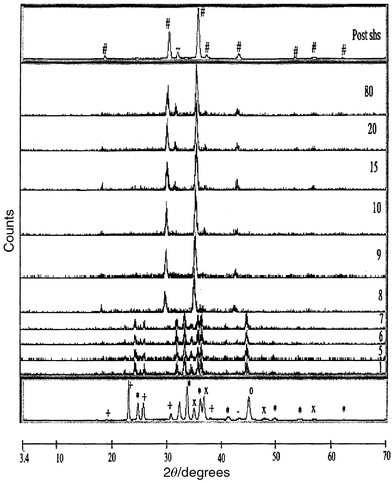 | ||
| Fig. 6 X-Ray powder diffraction patterns of the SHS reaction of ZnO, MgO, Fe, Fe2O3 and NaClO4 under a flow of oxygen. Patterns were obtained at 0.25 s intervals, the propagation front moves through the beam between run numbers 7 and 8. Plots are shown of intensity against 2θ. Key: + NaClO4; * Fe2O3; — MgO; × ZnO; # Mg0.5Zn0.5Fe2O4; ∼ NaCl. | ||
The d-spacings obtained for Mg0.5Zn0.5Fe2O4
are 1.1 to 1.3% larger on formation compared to the room temperature
post-SHS product. This gives an initial estimate of reaction temperature
in excess of 1800°C. The cooling rate in the magnesium zinc ferrite
system is also fairly rapid reaching 100°C 60 s after the
start of the reaction.
Lithium ferrite
°C by optical pyrometry
and 910°C by thermal imaging camera. The product is a brown powder.
Again like the barium ferrite and magnesium zinc ferrite reactions the green
mixture in the applied field lithium ferrite reactions was pulled into the
centre of the field to produce an hour glass shape in cross section. Applied
field reactions proceed with an orange coloured propagation wave of velocity
5 mm s−1. A temperature of 925°C
was measured by optical pyrometry, 1030°C by thermal imaging camera.
Shiny, fused parts were observed in the post-SHS product, although they
form less than 10% of the total mass.Homogeneity checks of the green mixture in the applied field reactions showed identical diffraction patterns measured at various distances from the magnet wall, indicating that on the macroscopic scale the starting material was homogeneous.
Li0.5Fe2.5O4 is observed as the main phase
in the post-SHS product in both the zero field and applied field reactions, eqn. (3), although traces of Fe2O3
and Fe were also present as minor phases. These phases are due to incomplete
reaction in the SHS stage; they disappear on sintering at 1150°C
for 6 h, leaving a single phase homogeneous product. The annealed and
as made samples had a cubic spinel structure and indexed well with literature
reports.12 The SEM of the annealed lithium
ferrites showed micron sized cubic crystallites. FT-IR analysis revealed
a broad band at 600 cm−1. The Mössbauer spectra
of Li0.5Fe2.5O4 showed two overlapping sextets
in a 3 ∶ 2 ratio corresponding to the octahedral and tetrahedral
iron sites respectively (Li0.5Fe2.5O4
has an inverse spinel structure with 3/5 of the Fe3+ in
octahedral sites and 2/5 of the Fe3+ in tetrahedral sites).
The Mössbauer parameters agreed well with previous literature measurements.
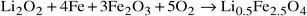 | (3) |
°C.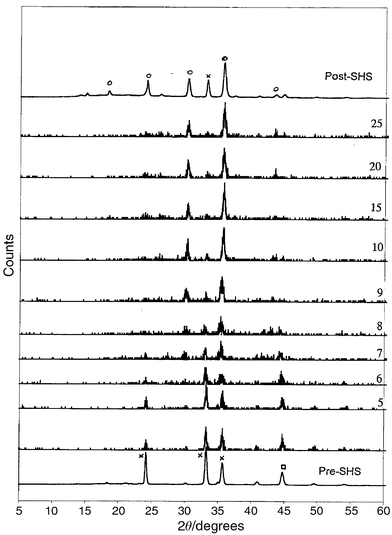 | ||
| Fig. 7 X-Ray powder diffraction stack plots of the SHS reaction of Li2O2, Fe, Fe2O3 under a flow of oxygen. Patterns were obtained at 0.1 s intervals. Plots are shown of intensity against 2θ. Key: × Fe2O3; □ Fe; ○ Li0.5Fe2.5O4. | ||
Discussion
Time resolved X-ray diffraction measurements on SHS reactions can be readily accomplished, using a white beam of X-rays and glancing angle detectors. This enables the study of very fast SHS reactions. With one notable exception the transformation from reactants to products in these SHS reactions is faster than the sampling rate used in the experiment. The transformation from the mixed starting material to crystalline product occurs faster than 250–100 ms. This is remarkably fast when compared to conventional solid state reactions which typically require multiple grinding, heating and cooling steps to reach completion. The major difference between these processes is that in SHS the energy for the transformation comes internally from within the reactant mixture and not from an external heating source. Hence aspects of heat transfer from a furnace or from the surroundings are negated. The SHS reaction proceeds by a synthesis wave that is hot enough to ignite successive layers of reactants. This process is extremely rapid.The only intermediate observed in these reactions was for the synthesis of barium ferrite in an applied magnetic field. The experiment was repeated five times to assess reproducibility. On four of these occasions a large diffraction peak that can be assigned to Fe3O4 was observed. The peak predominated in the spectra for 0.5 s. It is not possible at present to account for why this was seen in an applied field synthesis and not in the zero field experiments. One explanation could be that the propagation wave was parallel to the X-ray beam in the applied field synthesis but was perpendicular to the beam in the zero field experiments. Hence as the X-ray sampling ‘lozenge’ was ca. 0.1 mm wide but 4 mm long in the longitudinal direction, this may account for detection of intermediates. A further explanation is the fact that the applied field pre-organises the green mixture prior to the reaction and may give rise to a predominant Fe3O4 phase. Further experiments are planned to investigate these effects.
Previous work on time resolved X-ray powder diffraction studies of SHS reactions studied simple elemental combination reactions involving two or three component systems. These measurements were made on either conventional laboratory equipment over a very narrow 2θ range or on synchrotron sources using monochromatic X-rays and a bank of detectors that at most cover a 30° 2θ range. The experiments reported here are the first use of energy sensitive detectors and a white beam of X-rays to study SHS processes. The low incident angle of the X-ray beam and of the detector enabled the reactions to be studied inside a cylinder magnet, something that could not have been achieved in previous synchrotron experiments because the reflected X-rays would have been absorbed by the magnet before reaching the detectors. This is the first report of a TRXRD of a SHS reaction in an external magnetic field.
Previous TRXRD studies27–31 have shown that the SHS process is exceptionally swift; some tentative intermediates have also been observed in these reactions. The process of change from passage of the propagation wave to formation of products is of the order of 1–20 seconds dependent on the system. The ferrite oxide reactions studied here were essentially over within 1 s of the passage of the propagation wave. The extremely small spot size (0.1 mm) used in these experiments also meant that the confusion from overlapping products and reactant patterns was avoided.
In all of the systems studied there was a significant increase in the velocity and temperature of the synthesis wave with external field. In part this can be explained by the fact that in the presence of the external field all of the starting powder was pulled into the centre of the field to adopt an hour glass shape. Hence the geometries of the starting powders in the applied field and zero field syntheses were different. It is expected that the magnetic components, especially the iron metal, lined up in the field along field lines. This enables an easy propagation direction for the synthesis wave in the applied field cases. The oxidation of iron metal provides the fuel source for the reaction and pre-alignment enables easy heat transfer between neighbouring grains. This means that in the applied field reactions the process goes further to completion. This was definitely found to be the case for the barium ferrite reactions. In the lithium and magnesium zinc ferrite reactions the initial SHS reaction produces essentially the required ferrite phase in both the zero and applied field cases. In these systems the different synthesis wave temperatures observed are probably a consequence of the different shapes of the reactant powders and aspects of heat loss and transfer.
Some significant differences in the magnetism of the powders were observed between the zero and applied field syntheses, even after sintering at temperatures well in excess of the Curie temperature. It is probable that these changes in magnetism are related to differences of a microstructural origin. For example, in the barium ferrite synthesis the as formed product from the SHS reaction in an applied field showed lozenge shaped particles, whereas the zero field products were homogeneous.
Conclusions
Self-propagating high temperature synthesis provides an easy route to a range of ferrite magnets. The reactions proceed by a synthesis wave of temperature 800–1300°C and velocity 2–10 mm s−1,
dependent on the system. In some cases, such as lithium ferrite and magnesium
zinc ferrite, the desired ferrite is formed directly from the SHS, whilst
for hexagonal barium ferrite the product requires annealing at 1150°C
for 6 h. The materials synthesised by SHS have somewhat different magnetic
figures of merit to conventionally prepared ferrites. This can be traced in
some cases to a fine-grained microstructure. Time resolved X-ray powder
diffraction measurements of SHS reactions were obtained for the first time
using a white beam of X-rays and an energy sensitive detector. This system
enabled diffraction patterns to be measured at intervals as low as 100 ms.
TRXRD patterns were also obtained for the first time in an external magnetic
field.These studies have shown that in the ferrite systems studied the transformation
of reactants to products is complete in most cases in less than 100 ms.
This is extremely fast compared to conventional ceramic synthesis techniques.
The products as-formed in the SHS reactions show lattice expansion effects
due to the high formation temperatures. These expansion effects can also be
used to estimate rates of cooling within the system. This reveals that the
SHS process initially cools at a rate of ca. 1000°C min−1
for all three systems studied.
Further studies are planned for the use of TRXRD in fast chemical systems; it may be possible to record data in this set up with 50 ms or lower sampling rates.
Acknowledgements
MVK thanks both the Royal Society and the Royal Society of Chemistry Journal Fund for a short-term visitor grant which has facilitated this work. LA thanks the EPSRC for a Quota studentship. The Mössbauer spectra were collected under the auspices of the University of London Intercollegiate Research Service. We also thank Simon Clark of the CLRC Daresbury Laboratory for help in collecting data on station 16.4; Yuri Morozov (ISMAN, Moscow) is thanked for help with VSM measurements and for discussion and FLIR LTD thanked for the loan of a thermal-imaging camera.References
- H. Yanagida, K. Koumoto and M. Miguayuma, Chemistry of Ceramics, J. Wiley and Sons, Chicester, 1996, p. 97. Search PubMed.
- M. V. Cabañas, J. M. González-Cablet and M. Vallet-Regi, J. Solid State Chem., 1995, 115, 347 CrossRef CAS.
- D. H. Han, Z. Yang, H. X. Zeng, X. Z. Zhou and A. H. Morrish, J. Magn. Magn. Mater., 1994, 137, 191 CrossRef CAS.
- T. Ido, O. Kubo, H. Yokoyama and S. Kenjo, Toshiba Rev. Int. Ed., 1985, 154, 10 Search PubMed.
- T. Fujiwara, IEEE Trans. Magn., 1985, 21, 1480 CrossRef.
- A. G. Merzhanov, J. Mater. Proc. Technol., 1996, 56, 222 Search PubMed.
- A. G. Merzhanov, Adv. Mater., 1992, 4, 294 CrossRef.
- A. G. Merzhanov, Int. J. Self-Propag. High Temp. Synth., 1995, 4, 323 Search PubMed.
- Q. A. Pankhurst and I. P. Parkin, Mater. World, 1998, 743 Search PubMed.
- (a) I. P. Parkin, G. Elwin, L. Fernández-Barquín, Q. T. Bui, Q. A. Pankhurst, A. V. Komarov and Y. G. Morozov, Adv. Mater., 1997, 9, 643 CAS; (b) I. P. Parkin, M. V. Kuznetsov and Q. A. Pankhurst, J. Mater. Chem., 1999, 9, 273 RSC.
- M. V. Kuznetsov, Q. A. Pankhurst and I. P. Parkin, J. Mater. Chem., 1998, 8, 2701 RSC.
- M. V. Kuznetsov, Q. A. Pankhurst and I. P. Parkin, J. Phys. D, 1998, 31, 2886 Search PubMed.
- X. Z. Zhou, A. H. Morrish, Z. Yang and H. X. Xeng, J. Appl. Phys., 1994, 75, 5556 CrossRef CAS.
- L. Fernandez Barquin, M. V. Kuznetsov, Y. G. Morozov, Q. A. Pankhurst and I. P. Parkin, Int. J. Inorg. Mater., 1999, 1, 311 CrossRef.
- (a) A. Tomas, P. Laruelle, J. L. Dormann and M. Nouges, Acta Crystallogr., Sect. C, 1983, 39, 1615 CrossRef; (b) J. L. Dormann, Rev. Phys. Appl., 1980, 15, 1113 Search PubMed.
- W. B. Cross, L. Affleck, M. V. Kuznetsov, I. P. Parkin and Q. A. Pankhurst, J. Mater. Chem., 1999, 9, 2545 RSC.
- A. I. Kirdyashkin, Y. M. Maksimov and A. G. Merzhanov, Fizika Goreniya I Vzriva, 1986, 22, 65 Search PubMed.
- A. V. Komarov, Y. G. Morozov, P. B. Avakyan and M. D. Nersesyan, Int. J. Self-Propag. High-Temp. Synth., 1994, 3(3), 207 Search PubMed.
- M. D. Aguas, L. Affleck, I. P. Parkin, M. V. Kuznetsov, W. A. Steer, Q. A. Pankhurst, L. Fernandez Barquin, M. A. Roberts, M. Boamfa and J. A. A. Perenboom, J. Mater. Chem., 2000, 10, 235 RSC.
- M. D. Nersesyan, J. R. Claycomb, Q. Ming, J. H. Miller Jnr., J. T. Richardson and D. Luss, Appl. Phys. Lett., 1999, 75, 1170 CrossRef CAS.
- J. M. D. Coey, G. Hinds and M. E. G. Lyons, Europhys. Lett., 1999, 47, 267 Search PubMed.
- G. Hinds, J. M. D. Coey and M. E. G. Lyon, J. Appl. Phys., 1998, 83, 6447 CrossRef CAS.
- H. Yokomichi, H. Sakima, M. Ichihara, F. Sakai, K. Itoh and N. Kishimoto, Appl. Phys. Lett., 1999, 74, 1827 CrossRef CAS.
- Z. A. Munir and J. B. Holt (editors), Combustion and plasma synthesis of high temperature materials, VCH, New York, 1990, p. 1. Search PubMed.
- (a) C. R. Bowen and B. Derby, Acta Metall. Mater., 1995, 43, 3903 Search PubMed; (b) C. R. Bowen and B. Derby, Br. Ceram. Trans., 1997, 96, 25 Search PubMed.
- H. C. Yi and J. J. Moore, J. Mater. Sci., 1990, 25, 1159 CAS.
- E. M. Larson, J. Wong, J. B. Holt, P. A. Waide, G. Nutt, B. Rupp and L. J. Terminello, J. Mater. Res., 1993, 8, 1533 CAS.
- C. H. Gras, F. Charlot, E. Gaffet, F. Bernard and J. C. Niepce, Acta Mater., 1999, 47, 2113 CrossRef CAS.
- F. Charlot, F. Bernard, E. Gaffet, D. Klein and J. C. Niepce, Acta Mater., 1999, 47, 619 CrossRef CAS.
- J. F. Javel, M. Dirand, F. Z. Nassik and J. C. Gachon, J. Phys. IV, 1996, 6, C2229 Search PubMed.
- C. R. Kachelmyer, I. O. Khomenko, A. S. Rogachev and A. Varma, J. Mater. Res., 1997, 12, 3230 CAS.
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29 September 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |