Integrated chemical process for green chemistry. One-pot synthesis of 3-substituted 5-(β-sulfonylvinyl)indoles by in situ generation of toxic vinyl sulfone
Akihiro Oritaa, Masataka Katakamia, Yutaka Yasuia, Akio Kuriharab and Junzo Otera*a
aDepartment of Applied Chemistry, Okayama University of Science, Ridai-cho, Okayama, 700-0005, Japan.. E-mail: otera@high.ous.ac.jp
bOrganic Synthesis Research Laboratory, Sumitomo Chemical Co., Ltd., 10-1 2-Chome Tsukahara Takatsuki-City, Osaka, 569-1093, JapanFirst published as an Advance Article on the web
First published on 8th January 2001
Abstract
Integrated chemical processes where multi-step reactions are consolidated in one pot are effective for realizing a green chemical process. In situ generation followed by consumption of toxic intermediates which are formed in the process enables their complete isolation from the environment. This is so for a vinyl sulfone intermediate in the synthesis of 3-substituted 5-(β-sulfonylvinyl)indoles. The validity of this idea is further confirmed by successful one-pot operations on an automated synthesizer.
Green ContextMulti-step syntheses produce considerable amounts of waste, in part due to their complexity, but also due to a series of complex isolation procedures which must be carried out after each step. Thus, the combination of steps into a multi-step, one-pot reaction sequence can be of great value, as long as overall yield and efficiency are not negatively influenced. The chemistry described in this article is a good example of an efficient process which has been carried out over several steps without isolation of intermediates, and with excellent efficiency. Additionally, some of the toxic intermediates formed are not isolated, and thus Health and Safety aspects of the chemistry are improved.DJM |
Introduction
Previously, we introduced a new concept for consolidation of multi-step reactions in one-pot, a so-called ‘integrated chemical process’. In this treatment, reaction conditions which can be tolerated by all steps are established first and, then, each step is optimized within the framework of these conditions.1 We have already disclosed that such a procedure not only effects compaction of the whole process but also gives rise to an increase in overall yield. Moreover, another advantage lies in the enclosure of relevant intermediates in the reaction flask throughout the process. This may be of great promise in terms of green chemistry because processes involving toxic intermediates can be operated without exposure of these substances to the environment.3-Substituted 5-(β-sulfonylvinyl)indoles are useful intermediates for a variety of pharmaceuticals and are fabricated by Heck reaction between vinyl sulfone and the corresponding 5-halo indoles.2 Phenyl vinyl sulfone, though being a versatile reagent which is used in a wide spectrum of organic syntheses,3 is an irritant to the eyes, the respiratory system and skin on account of their facile susceptibility to the Michael reaction4 and, thus, handling a mass of the neat compound in the open air must be avoided. We demonstrate here that the problem can be overcome by integrating two steps consisting of phenyl vinyl sulfone formation from non-toxic 2-sulfonylethanol and subsequent Heck reaction with 5-bromoindole.
Results and discussion
Initially, we assessed the validity of our concept by comparing stepwise and integrated processes using model reactions [eqns. (1) and (2)]. The preparation of phenyl vinyl sulfone 2 | (1) |
 | (2) |

Next, both steps were brought together. As expected, the two reactions were virtually compatible but the overall yields were considerably dependent on the conditions for the first step. Among the procedures described above for 2, the mesylation method gave the best outcome when aryl iodides were the reaction components in the Heck reaction. In this process, acetonitrile was the solvent of choice. In Table 3, the overall yields thus obtained are shown together with those via the stepwise process for comparison (entries 1–6). On the other hand, trifluoroacetic anhydride (TFAA) gave better yields than the other reagents when 5-bromo- or 5-iodo-indole was subjected to the Heck reaction (entries 7–10). DMF was the common solvent for the consecutive reactions. Remarkably, the integrated protocol provided higher yields (except for entry 4) than the corresponding stepwise process regardless of the method for the generation of 2.

With these results in hand, we turned out attention on the one-pot synthesis of 5-(E)-(2-phenylsulfonylvinyl)-3-(N-methylpyrrolidin-2-yl methyl)-1H-indole 4, which, upon reduction of the vinyl moiety, is converted to the corresponding 5-(2-phenylsulfonylethyl) derivative, an intermediate for drug compositions effective for treating migraine and other disorders.2b As shown Scheme 1, the vinyl sulfone 2 was generated by the TFAA method and subsequently treated with 5-bromo-3-(N-methylpyrrolidin-2-ylmethyl)-1H-indole 5. When the Heck reaction was complete, a trace amount of unreacted 2 was detected in the reaction mixture by HPLC monitoring. The addition of Et2NH completely consumed 2 without destruction of 4. As a result, the phenyl vinyl sulfone was not exposed to the environment during the entire manipulation process. The overall yield was 58% on the basis of HPLC analysis and a 56% yield was obtained by preparative HPLC. However, the isolated yield was somewhat decreased (51%) because of the instability of 4, yet remarkably higher than that of the stepwise process (38% = 74% × 51%). This may be attributed to the efficiency in generation of 2 as well as in its use for the Heck reaction. Essentially, the integrated process can minimize the mechanical loss of the intermediates during manipulations for isolation and purification. Furthermore the vinyl sulfone itself is not very chemically stable5 and, thus, the stepwise process suffers a serious decrease in the overall yield. It is also rather surprising that, despite the instability of 4, a reasonable yield was accessible from the reaction mixture which contained various materials resulting from the two reactions, indicative of the practical usefulness of the integrated chemical process.
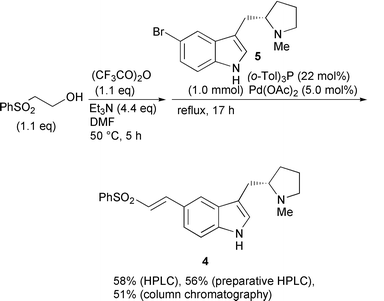 | ||
| Scheme 1 One-pot synthesis of 4. | ||
Finally, the validity of the present protocol as a green process was further proved by running it on an automated synthesizer (MEDLEY) which we have recently developed.6 This machine enables successive addition of reagents in precise amounts (within ±0.01 mL) and control of the reaction temperature to ±0.1 °C. More importantly, the reaction can be run under a completely inert atmosphere to accommodate air-sensitive organometallic or transition metal compounds. We thus carried out the automated synthesis of 4 on MEDLEY. As shown in Scheme 2, a DMF solution of 1 was placed in a reaction flask and then the program was initiated. Nine operations were performed automatically and 4 was obtained in 58% yield based on HPLC analysis. This integrated chemical process is clearly environmentally benign.
 | ||
| Scheme 2 One-pot synthesis of 4 on MEDLEY. | ||
Conclusion
An integrated chemical process has proved to be effective for realizing a green process, enabling isolation of a toxic intermediate from the environment through in situ generation followed by consumption. There are a number of processes in which toxic intermediates are inevitably generated and, accordingly, the concept disclosed here will find diverse applications.Experimental
General comments
All reactions were carried out under nitrogen unless otherwise noted. Acetonitrile, dimethylformamide and triethylamine were distilled from calcium hydride. NMR spectra were recorded at 25 °C on JEOL Lambda 300 and 500 instruments and calibrated with tetramethylsilane (TMS) as an internal reference. High-performance liquid chromatography (HPLC) was performed on a Recycling Preparative HPLC LC-908 (Japan Analytical Industy Co., Ltd) equipped with GPC columns JAIGEL-1H and 2H using refractometer and UV detectors. Mass spectra were recorded on a JEOL MStation JMS-700 spectrometer. Elemental analyses were performed on a Perkin Elmer PE 2400 instrument. 2-Phenylsulfonylethanol 1 was provided by Sumitomo Chemical Co., Ltd. Silica gel (Daiso gel IR-60) was used for column chromatography.Representative preparations
Characterization data
Preparation of 5-(E)-(2-phenylsulfonylvinyl)-3-(N-methyl- pyrrolidin-2- ylmethyl)-1H-indole 4 in a one-pot procedure
To a flame-dried flask were added 2 (204 mg, 1.1 mmol), trifluoroacetic anhydride (0.17 mL, 1.1 mmol), triethylamine (0.62 mL, 4.4 mmol) and DMF (1.0 mL), and the mixture was heated at 50 °C for 5 h. After confirmation of complete consumption of 2 by TLC monitoring, a DMF solution (1.0 mL) of palladium acetate (11 mg, 0.05 mmol) and tris(o-tolyl)phosphine (67 mg, 0.22 mmol) was added. Then, a DMF solution (1.0 mL) of 5 (293 mg, 1.0 mmol) was added and the mixture was heated under reflux for 17 h with the flask shielded from light. Et2NH was added (0.016 mL) and the mixture was stirred at room temperature until the vinyl sulfone disappeared (HPLC monitoring). After addition of water and extraction with ethyl acetate, the organic layer was washed with brine, dried over Na2SO4 and evaporated. HPLC analysis of the crude mixture indicated a 58% yield of 4. Preparative HPLC afforded 213 mg (56%) of 4 while column chromatography on silica gel [methanol–chloroform–conc. NH3(aq) (50∶50∶1) → dichloromethane–methanol–conc. NH3(aq) (90∶10∶1) afforded 194 mg (51% yield): δH(CDCl3) 1.50–1.90 (m, 4H), 2.24 (q, J 8.2 Hz, 1H), 2.37–2.53 (m, 1H), 2.46 (s, 3H), 2.54–2.67 (m, 1H), 3.10–3.20 (m, 2H), 6.79 (d, J 15.3 Hz, 1H), 7.07 (s, 1H), 7.30–7.40 (m, 2H), 7.51–7.65 (m, 3H), 7.74 (s, 1H), 7.84 (d, J 15.3 Hz, 1H), 7.98 (d, J 7.9 Hz, 2H), 8.26 (br, 1H); δC(CDCl3) δ 21.8, 29.7, 31.4, 40.8, 57.5, 66.5, 111.9, 115.2, 121.5, 121.8, 123.2, 123.4, 123.7, 127.4, 128.0, 133.0, 137.8, 141.4, 144.7.Preparation of 5-(E)-(2-phenylsulfonylvinyl)-3-(N-methyl- pyrrolidin-2-ylmethyl)-1H-indole 4 on MEDLEY
A flask equipped with a reflux condenser was attached to MEDLEY and flame-dried under reduced pressure. Under a stream of nitrogen, 2 was added to the flask, and the flask was placed in an oil bath. To the storage tanks of MEDLEY were added trifluoroacetic anhydride, triethylamine, a DMF solution of 5, a THF solution of Pd(OAc)2 and tris(o-tolyl)phosphine and a DMF solution of Et2NH. The reaction temperatures and periods shown in Scheme 2 were programmed, and the operations started. After the completion of the programmed operations, water was added and the aqueous layer was extracted with ethyl acetate. The organic layer was washed with brine. After drying over Na2SO4 and evaporation, the crude mixture was filtered through a silica gel pad [methanol–chloroform–conc. NH3(aq) 50∶50∶1) → dichloromethane–methanol–conc. NH3(aq) (90∶10∶1)] and HPLC separation afforded a pure product (419 mg, 55% yield).Acknowledgements
Thanks are due to Dr Tadatoshi Aratani of Sumitomo Chemical Co. Ltd. for his helpful discussion while assistance in experimental work by H. Watanabe is gratefully acknowledged.References
- (a) A. Orita, Y. Yamashita, A. Toh and J. Otera, Angew. Chem., Int. Ed. Engl., 1997, 36, 779 CrossRef CAS; (b) A. Orita, N. Yoshioka and J. Otera, Chem. Lett., 1997, 1023 CrossRef CAS; (c) A. Orita, A. Watanabe and J. Otera, Chem. Lett., 1997, 1025 CrossRef CAS; (d) A. Orita, A. Watanabe, H. Tsuchiya and J. Otera, Tetrahedron, 1999, 55, 2889 CrossRef CAS; (e) A. Orita, N. Yoshioka, P. Struwe, A. Braier, A. Beckmann and J. Otera, Chem. Eur. J., 1999, 5, 1355 CrossRef CAS; (f) A. Orita, J. Yaruva and J. Otera, Angew. Chem., Int. Ed., 1999, 38, 2267 CrossRef.
- (a) P. C. North, M. R. Johnson and A. W. Oxford, Eur. Pat. Appl., 90301419.9, 1990;; (b) J. E. Macor and M. J. Wythes, US Pat., 5 545 644, 1996..
- N. S. Simpkins, Sulphones in Organic Synthesis, Pergamon Press, Oxford, 1993, ch. 4. Search PubMed.
- (a) S. C. DeVito, CHEMTECH, 1996, November, 34;Aldrich Material Safety Data Sheet, Product # 79293..
- After the preparation of 2 [eqn. (1)], TLC monitoring exhibited a single spot attributable to the desired product, however, isolation by column chromatography always gave rise to less than 75% yields suggesting partial decomposition of 2..
- A. Orita, Y. Yasui and J. Otera, Org. Process Res. Dev., in press. Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |