Partitioning of mercury onto suspended sediments in estuaries†
Sophie M.
Le Roux
*ab,
Andrew
Turner
a,
Geoffrey E.
Millward
a,
Les
Ebdon
a and
Pierre
Appriou
b
aDepartment of Environmental
Sciences, University of Plymouth, Drake Circus, Plymouth, UK PL4 8AA. E-mail: sleroux@plymouth.ac.uk; Fax: +44 (0) 1752 233035; Tel: +44 (0)
1752 233022
bLaboratoire de Chimie Analytique Marine, Département de Chimie, UMR CNRS 6521, Université de Bretagne Occidentale, 6 avenue Le Gorgeu, 29285, Brest
Cedex, France
First published on 13th December 2000
Abstract
Radiochemical partitioning experiments using 203Hg have been undertaken with mixtures of river, seawater and sediment samples taken from three geochemically contrasting UK estuaries: the Plym, Beaulieu and Mersey. Species of dissolved Hg were determined using reversed-phase C18 chelating columns and particulate species were determined by sequential leaching with 1 M NH4OAc and 1 M HCl. Mercury had a high particle reactivity with partition coefficients, KDs, ranging from 104 to 5 × 105 ml g−1, depending on salinity, the chemical composition of the end-member waters, and on the physico-chemical characteristics of the sediment. Dissolved organic matter present in the waters (humic substances and/or anthropogenic compounds) was found to be the main factor governing the forms of dissolved Hg and their reactivity. From the spiked 203Hg, up to 95% of the dissolved metal was retained on the C18 columns for the Mersey waters, whereas this fraction was <60% in the Plym and Beaulieu waters. Quasi-irreversible adsorption of Hg onto particles from each estuary was observed over a time-scale of a few hours and <20% of total particulate Hg was released by the sequential leach. In this paper, physico-chemical processes are proposed to explain the estuarine behaviour of Hg and the results are discussed in terms of Hg availability in estuarine systems.
Introduction
The behaviour and speciation of metals in estuaries play a major role in their biogeochemical cycling,1,2 an understanding of which is essential in order to assess the impact of metal pollution on coastal and marine ecosystems. In particular, contamination of the environment by Hg is a major global issue3,4 owing to the acute toxicity of this element. Recent advances in analytical techniques5 have enabled accurate measurements of low concentrations of various Hg species encountered in aquatic systems. A number of intensive investigations have been devoted to assessing Hg contamination and bioaccumulation in polluted sites.6–10 However, there remains an incomplete understanding of the factors controlling the speciation and particle–water reactivity of Hg in estuaries, where reaction conditions change markedly on going from river to sea.The work presented in this paper has attempted to address this question by controlled laboratory experiments in which the γ-emitting radioisotope 203Hg (t1/2 = 47 days) was used as a tracer. The experiments were based on a simulation of estuarine processes, using salinity as an indicator of mixing. Water and sediment samples were obtained from three UK estuaries with contrasting geochemical properties, such as dissolved organic carbon (DOC), suspended particulate matter (SPM) concentrations and the type of SPM. The radiochemical partitioning experiments replicated instantaneous particle–water interactions, and therefore allow direct insight into trace-metal estuarine reactivity.11,12 The advantages of using radioisotopes to simulate the behaviour of their stable analogues are: (a) the possibility of working at ultra-trace levels (pico- to nanomolar) offered by the low limits of detection of γ-spectrometry; (b) the elimination of contamination problems; and (c) the direct analysis of particulate and dissolved samples.13 A novel approach was used in this work to complement the partitioning experiments, by applying trace-metal speciation protocols to the 203Hg radioisotope in both the dissolved and the particulate phases. The experiments were designed to answer the following questions: (1) What are the physico-chemical factors controlling the speciation of dissolved Hg in estuarine waters? (2) What processes control the particle–water interactions of Hg in estuaries? (3) What are the environmental implications of Hg reactivity in estuarine systems?
Methodology
Sampling, sample treatment and analysis were performed using ultra-clean protocols. Ultra-pure water was obtained from a Millipore (Watford, Hertfordshire, UK) Milli-Q/Milli-Ro system (18.2 MΩ resistivity). All glassware and plasticware used in the radiochemical experiments were previously soaked for a least 24 h in a bath containing 10% Decon (Merck BDH, Poole, Dorset, UK), then in 3 M HCl and finally thoroughly rinsed with ultra-pure water. All reagents used were AnalaR or AristaR grade (Merck BDH). When possible, sample handling was performed in a laminar flow hood.Sampling
| Plym (May 1998) | Mersey 1 (June 1998) | Mersey 2 (October 1998) | Beaulieu (March 1998) | ||
|---|---|---|---|---|---|
a DOC: dissolved organic
carbon; POC: particulate organic carbon; SSA: specific surface area.
b ND: not determined.
c Total mineralisation
using HNO3 + HCl + HF (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C).
d Average and standard deviation of three replicates. °C).
d Average and standard deviation of three replicates.
|
|||||
| River water | pH | 6.74 | 7.03 | 7.39 | 6.93 |
| DOC/mg C l−1 | 1–3 | 9.7 | 9.33 | 17.7 | |
| Conductivity/mS | 0.5 | 0.63 | 0.38 | 0.27 | |
| Seawater | PH | 8.11 | 8.07 | 7.88 | 8.09 |
| DOC/mg C l−1 | 1–2 | 1.8 | 1.9 | 1.6 | |
| Salinity | 32.8 | 32.5 | 31.2 | 33.9 | |
| Sediment (<63 µm) | SSA/m2 g−1 | 6.1 | 9.3 | 6.0 | 6.9 |
| POC (%) | 2.1 | 7.5 | NDb | 6.9 | |
| Total metalc | |||||
| Ald/mg g−1 | 69.2 ± 1.4 | 64.0 ± 2.6 | 14.0 ± 1.5 | 33.7 ± 2.7 | |
| Fed/mg g−1 | 25.0 ± 0.4 | 48.0 ± 2.1 | 8.27 ± 0.9 | 36.7 ± 2.9 | |
| Mnd/mg g−1 | 0.53 ± 0.25 | 1.20 ± 0.32 | 1.19 ± 0.13 | <0.28 | |
| Fe ∶ Al | 0.36 | 0.75 | 0.59 | 1.09 | |
| Mn ∶ Al × 103 | 7.7 | 19 | 85 | <8.3 | |
The River Plym drains a moorland catchment area with granitic rock and various soil types. The estuary is characterised by relatively low DOC and SPM concentrations (Table 1). By contrast, the River Beaulieu exhibits elevated DOC concentrations (Table 1), since it drains an afforested and moorland catchment area. Whereas these two estuaries possess relatively ‘clean’ drainage basins, the Mersey estuary is located in an intense industrial area. The river drains the largest urban area in the UK and the estuary is, or has been, subject to untreated discharges of domestic sewage and industrial wastes.14,15 Water and sediment contamination of the estuary includes a variety of partially identified organic compounds such as surfactants and organochlorines, and inorganic pollutants such as heavy metals, nitrogen and sulfides. In particular, Hg contamination still remains a major concern due to previous indirect discharges in dissolved and particulate form from chloroalkali plants and paper mills.16
Freshwater samples were collected from the shore at the limit of the saline
intrusion and seawater samples offshore at the mouth of the estuary, using
acid-cleaned 5 l Nalgene (Nalge Ltd, Hereford, UK) bottles.
The samples were filtered within 24 h of collection through acid-cleaned
0.45 µm pore size cellulose acetate membranes (47 mm
diameter, Millipore) using a Nalgene filtration kit, and stored at 4°C.
For DOC analyses, additional water samples were collected in 250 ml
borosilicate glass bottles previously heated at 550°C for 8 h.
These samples were filtered through ashed 0.7 µm pore size GF/C
membranes (47 mm diameter, Whatman, International Ltd, Maidstone,
Kent, UK) using a glass filtration unit. After addition of 0.3%
v/v H3PO4, the filtrates were analysed for DOC using
a Shimadzu (Milton Keynes, UK) TOC-5000 total organic carbon
analyser. Other physico-chemical parameters were measured during sampling, i.e.,
pH, salinity and temperature, dissolved oxygen and conductivity.
Surface estuarine sediments from the Plym and Beaulieu estuaries were collected
manually from inter-tidal mud using a PTFE spatula, whereas sediment samples
from the Mersey were collected at the same location as the river water using
a Shipek grab. The sediments were separated across an acid-cleaned 63 µm
nylon sieve using a small quantity of the native river water and stored at
4°C until required for the incubation experiments. A few grams
of the <63 µm sediment fraction were dried in a laminar flow
hood at room temperature then at 60°C overnight, ground in a mortar
and stored in a vacuum desiccator. Total iron, aluminium and manganese concentrations
were determined by GFAAS in these samples after HF mineralisation following
a protocol described by Rantala and Loring.17
The dried sediments were also used for specific surface area (SSA)
measurements using a N2 gas adsorption method in a Micromeritics (Micromeritics
Instrument Corporation, Norcross, GA, USA) Gemini 2360 analyser. Sediments
for particulate organic carbon (POC) analysis were stored in sterilised
borosilicate bottles, then separated across a 63 µm sieve and
filtered through 0.7 µm pore size GF/C membranes (47 mm
diameter; Whatman). After drying at room temperature, the samples were
ground in an agate mortar and determined combustimetrically using a Shimadzu
TOC-5000 CHN analyser.
Radiochemical experiments
The overall protocol for partitioning and speciation of 203Hg is shown in Fig. 1. | ||
| Fig. 1 Schematic flow chart of the radiochemical partitioning experiments, and of dissolved and particulate speciation procedures. | ||
°C. A 20 ml aliquot was then pipetted
from each incubation reactor into 20 ml scintillation vials for γ-counting (A0).
Another 20 ml collected from the reactors were passed through C18
columns in order to isolate hydrophobic complexes formed with the added isotope,
following the protocol described below. To the solutions remaining in the
incubation bottles, a constant amount of sieved estuarine sediment was then
added to reach a final concentration of approximately 100 mg l−1
in each reactor. Preliminary incubation experiments using various proportions
of suspended solid and added 203Hg showed that this solid ∶ Hg
ratio was too high for any saturation of the particle binding sites to occur.
Following equilibration for 24 h, under mechanical shaking, at 20°C
and in the dark, the particulate and dissolved fractions were separated by
filtration through pre-weighed 0.45 µm pore size cellulose
acetate membranes (22 mm diameter; Sartorius AG, Göttingen,
Germany). Preliminary kinetic studies showed that 24 h of incubation
were sufficient for the partitioning process to reach equilibrium, due to
the high particulate reactivity of mercury. The filters and 20 ml of
the filtered solutions were then placed into scintillation vials to measure
particulate and dissolved 203Hg activity, AP
and AS, respectively (Fig. 1).
The filters were dried at room temperature and weighed for SPM determinations,
and then each filter was sequentially leached following the procedure described
below.
Gamma-energies (279 keV) were counted using a high performance Wallac 1480 Wizard 3″ γ-counter with NaI detection (Perkin-Elmer, Cambridge, UK); each sample was counted three times for 1000 s and automatically background corrected. Given the relative length of the overall experiments and the relatively short half-life of 203Hg, all activities were corrected for radioactive decay.
Mercury partition coefficients, KDs (ml g−1), were calculated as follows:11,12
 | (1) |
Validation of the C18 speciation scheme was performed using breakthrough and elution studies of five replicates. Saturation of the C18 columns was not observed after passing through 20 ml of spiked solutions. γ-counting of 2 ml fractions eluted by a total volume of 10 ml HNO3 revealed that 90% of the total eluted 203Hg was found in the first 2 ml of eluate collected. However, recoveries calculated from the initial activity, A0 (Fig. 1), and the sum of the activities at the end of the columns, AC, and in the eluate, AE, were only 30% on average. The Hg-C18 fraction, operationally defined as the organically bound mercury, was therefore calculated from the difference between the activities at the top and at the end of the columns (A0 − AC). This fraction includes all the metal retained on the columns, either by chelation between mercury hydrophobic complexes and the C18 functional groups, or by adsorption of 203Hg onto the columns. AE was used to calculate the eluted and resistant to 2 M HNO3 fractions. The Hg-C18 percentages given have a relative standard deviation of 6% (n = 5).
Recoveries of particulate mercury after the partitioning experiments were calculated from the initial activity on the filters and the sum of the activities on the filters and in the leachates after each extraction. Reproducibility of the procedure was better than ±3% (n = 5), with an average 203Hg recovery of 95 ± 3%.
Results and discussion
Sample characteristics
Physico-chemical characteristics of the samples used in the partitioning experiments are given in Table 1. The diversity of the catchment area between the three estuaries was reflected by the contrasting organic contents of the river water samples collected, with DOC concentrations ranging from 1 mg C l−1 in the Plym river to 17.7 mg C l−1 in the Beaulieu. Intermediate DOC concentrations were found in the Mersey river for both June and October surveys.The estuarine sediment samples collected also exhibited varied characteristics. The Mersey particles from the June survey possessed the highest POC, SSA and total iron and manganese concentrations (Table 1). These three parameters are interrelated, associations of Fe/Mn oxides and organic matter forming coatings at the surface of particles; they are thus an important criterion for particle surface reactivity.2,21 Aluminium normalisation can be used to assess the importance of clays and alumino-silicates with regard to metal sorption compared to other reactive phases, such as ferromanganese oxides and POC.22 The Mn ∶ Al ratios were higher in the Mersey sediments than in particles from the other estuaries, again suggesting their higher surface reactivity due to the presence of Mn oxides.
Partitioning and speciation of mercury on estuarine mixing
From thermodynamic modelling, Hg can be characterised by highly stable soluble complexes, either by binding with humic materials in fresh waters,23,24 or by complexation with seawater chloride ions to form mainly the four species HgCl+, HgCl2, HgCl3− and HgCl42−.25 However, whereas a predominance of dissolved forms of Hg would be expected from these predictive calculations, Hg partitioning in the three estuaries studied was dominated by its high particle reactivity, with KD > 104 ml g−1 in all cases, as shown in Fig. 2. Previous experiments conducted with the 203Hg radioisotope similarly provided typical KDs around 105 ml g−1.10,26–28 Another feature of Hg partitioning was the general increase of KD with increasing salinity (Fig. 2), despite the stability of chlorocomplexes. Although the relative standard deviation on the KD values in seawater was as high as 30% in the Plym and the Beaulieu, due to low 203Hg counts in solution, the increase of KD with salinity was significant.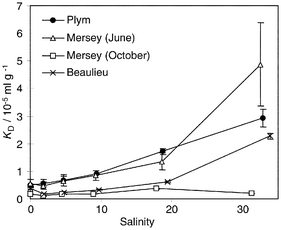 | ||
| Fig. 2 Mercury partition coefficients (KDs) in the Plym, Mersey and Beaulieu estuaries as a function of the salinity; error bars represent standard deviation of five replicates. | ||
The apparent contradiction between the stability of dissolved Hg complexes and its strong sorptive behaviour may be explained by the presence of the thermodynamically favoured HgOH+ hydrolysed species in oxic waters and in the range of pH considered.11 Hg hydrolysis competes with the formation of other stable complexes such as chlorocomplexes and is enhanced at higher pHs, providing one possible explanation for the increased KDs in seawater for the Plym, Beaulieu and Mersey (June) estuaries (Fig. 2). No increase was observed with the October Mersey samples, possibly due to the small pH difference between the river and seawater samples collected (Table 1).
In addition, mercury rich colloids can be formed either by coagulation and aggregation of Hg hydrolysed species or by metal sorption onto existing colloids. This ‘colloidal pumping’, evidenced by Stordal et al.27 using 203Hg incubation experiments, is particularly effective in an estuarine environment where the increase of ionic strength can cause flocculation of macromolecular organic matter and associated metals,29 and thus their transfer into the particulate phase.
The speciation of dissolved Hg performed through the use of reversed-phase C18 columns confirmed its affinity for hydrophobic organic compounds, with the Hg-C18 fractions exceeding 40% of the total dissolved Hg in the waters of the three estuaries studied, up to a salinity of approximately 20 (Fig. 3). The Hg-C18 fraction significantly decreased with increasing salinity for all estuaries, as the result of chlorocomplexation in seawater. However, the DOC concentrations of the waters could not be a factor explaining the variability of Hg–organic complexation, for three reasons. Firstly, a marked decrease of Hg-C18 from 50% in the river to less than 1% in seawater was observed in the Plym estuary, but the difference in DOC concentrations between the fresh and seawaters in this estuary (Table 1) was too low to account for this decrease. Secondly, the Hg-C18 percentages in fresh water did not follow the order of the organic contents of the river waters, i.e., Beaulieu > Mersey > Plym (Table 1). Thirdly, in spite of the similar DOC values in the June and October Mersey waters, a 30% difference in average was observed between the Hg-C18 fractions of the two surveys; the curves otherwise followed the same trend along the salinity gradient (Fig. 3). For these reasons, the type of DOC present in the waters appeared to be the factor dominating dissolved Hg speciation, rather than purely DOC concentrations. It appears that organic complexes formed with a natural form of organic matter, such as humic acids, were less hydrophobic than organic complexes formed in the waters of the Mersey estuary, where the natural DOC may be mixed with anthropogenic organic compounds. Indeed, the Hg-C18 fraction was generally lower in the Plym and Beaulieu waters than in those from the Mersey, where up to 95% of mercury was retained on the C18 columns in the October survey. In addition, the organic complexes formed in the Mersey waters were more strongly bound to the C18 columns, since less than 4% could be eluted by 2 M HNO3 (Fig. 3). This resistant fraction was on average 30% in the Beaulieu estuary and 40% in the Plym.
 | ||
| Fig. 3 Percentage of Hg-C18 in filtered (<0.45 µm) estuarine waters as a function of salinity: A, Plym; B, Mersey (June); C, Mersey (October); and D, Beaulieu; dark and light grey shades represent, respectively, the eluted by 2 M HNO3 and resistant fractions; error bars represent standard deviation of five replicates. | ||
The role of natural organic matter (NOM)
Solid/solution partitioning of Hg appeared related to its organic complexation, i.e., to the hydrophobic character of the complexes formed, and to their behaviour in the salinity gradient. Firstly, less organically bound Hg in seawater (Fig. 3) corresponded to an increased sorption onto the particles (Fig. 2). Furthermore, in the Mersey waters in October, where the highest organic complexation was found, the KDs were the lowest, below 4 × 104 ml g−1, and constant at all salinities. Mercury solubility could therefore be explained by competitive processes between its organic binding in solution, chlorocomplexation and particle sorption. The increased Hg sorption onto the particles from a salinity of 20 may be due to the adsorption of organic complexes onto the particles.7,28,30 Indeed, whereas adsorption of NOM and associated metals onto solid surfaces was expected to decrease with increasing pH,1 in seawater this trend may have been reversed by NOM salting out at high salinity and high ionic strength. The ‘salting out’ process, described by Turner and Rawling,31 arises in seawater because less water molecules are present to stabilise NOM in solution due to their association with the hydration sphere of dissociated seawater ions, and because of the compression of surface electrical double layer at high salinity.However, it should be noted that Hg complexing capacities were not measured in this study. If any saturation of the waters complexing capacity by the added 203Hg occurred, dissolved Hg speciation and its binding to particles would have been greatly affected.32 The diminution of Hg complexing capacity with salinity could also be a factor explaining the increase of KDs.
The increased sorption of Hg with salinity was observed in all cases except in the Mersey in October (Fig. 2). Similar studies33 also carried out on Mersey waters in June and October 1998 showed the same type of Hg distributions, and KDs higher in June than in October. No correlation with biological processes was found, although a dependency of Hg species to seasonal variations was established in other aquatic systems.34,35
Mercury particle reactivity
The rapid, almost irreversible adsorption of Hg onto particles in aquatic systems has been demonstrated in a number of in situ4,6,16,35 and laboratory11,27,28 studies. In this work, the high particle reactivity of Hg resulted in elevated KDs (Fig. 2), and additional evidence was given by chemical extractions performed on the 203Hg incubated sediments. After only 24 h of incubation, more than 80% of total particulate mercury was found irreversibly bound to the solids and could not be released even after extraction with 1 M HCl (Fig. 4). | ||
| Fig. 4 Distribution of particulate mercury between the exchangeable, leachable and residual fractions in estuarine sediments: A, Plym; B, Mersey (June); C, Mersey (October); and D, Beaulieu as a function of salinity. | ||
Several processes can be proposed to explain 203Hg irreversible migration into the solid lattice: sorption on high energy binding sites, isotopic exchange with Hg already present in the original particles, or formation of stable associations with particulate organic compounds.7,28,36 Mercury may also bind ferromanganese oxides by surface sorption or co-precipitation and then become refractory by ageing of the oxides.8,23,35,37,38 Mercury surface precipitation39 was observed in our laboratory at nanomolar Hg concentrations, using incubation experiments of the 203Hg radiotracer with several synthetic Fe and Mn oxides at various pHs. Another factor limiting Hg bioavailability in oxic waters may be the presence of metal sulfide phases in the particles. Recent research has shown that sulfide complexes are stable in saline waters,40,41 and exchange reactions between Hg and other metals present in these phases are likely to influence Hg partitioning.
Our data suggested no salinity-induced desorption of mercury from the particles (Fig. 4), in accordance with the results from Jones42 who found very little desorption from natural Hg-contaminated sediments subjected to seawater over a 7 day period. This is consistent with the observation that Hg sorption onto the particles generally increased with salinity (Fig. 2). The difference in Hg partitioning between the two Mersey campaigns was attributed to the variability of the organic compounds present in the estuary. The particle characteristics were another parameter that had to be taken into account. The particles collected in June exhibited a higher total Fe concentration (48 mg g−1 compared to 8.3 mg g−1 in October) and a higher SSA (9.3 m2 g−1 compared to 6.0 m2 g−1 in October; Table 1). Their higher surface reactivity could therefore account for the 3- to 10-fold higher KD values in June than in October. The higher reactivity of the Mersey particles compared to those from the two other estuaries was also reflected by the significantly lower Hg availability from these particles, i.e., the exchangeable and leachable fractions (Fig. 4).
The predominance of Hg in a refractory form in the particles (>80%) has important implications for the fate of Hg in the estuarine environment. From our results, Hg solubility and bioavailability would be severely limited in natural waters, as the majority of the metal was found in a refractory form unlikely to be released from the particles. The long-term consequences of such a particle reactivity would be the immobilisation and accumulation of Hg in estuarine sediments. However, surface sediments can be a major source of bioavailable Hg by biologically mediated methylation.10,24,35 In estuaries, although the methylation rate diminishes at increased pH and salinity,7 inputs of inorganic or methylated Hg into the water column can occur following sediment resuspensions, particularly in macrotidal estuaries such as the Plym, Beaulieu and Mersey.
Acknowledgements
S.L.R. is very grateful to the Whistler 2000 Speciation Symposium organisers for the travel bursary awarded for her attendance at the conference. Many thanks are due to the following persons: Dr. P. D. Jones (Environment Agency for England and Wales) and Miss S. Watts (University of Plymouth) for assistance with sampling on the Mersey estuary, Mr D. Henon (University of Plymouth) for the HF digests, and Dr. A. R. Bowie (University of Plymouth) for proof-reading the final manuscript.References
- A. C. M. Bourg, Cont. Shelf Res., 1987, 7, 1319 CrossRef.
- G. E. Millward, Analyst, 1995, 20, 609 RSC.
- World Health Organisation, Environmental Criteria 101: Methylmercury, World Health Organisation, Geneva, Switzerland, 1990. Search PubMed.
- R. J. M. Hudson, S.A. Gherini, W.F. Fitzgerald and D.B. Porcella, Water, Air, Soil Pollut., 1995, 80, 265 CAS.
- Ph. Quevauviller, M. Filippelli and M. Horvat, Trends Anal. Chem., 2000, 19, 157 CrossRef CAS.
- D. Airey and P. D. Jones, Water Res., 1982, 16, 577 CrossRef.
- N. Mikac, S. Niessen, B. Ouddane and M. Wartel, Appl. Organomet. Chem., 1999, 13, 715 CrossRef CAS.
- B. Quémerais, D. Cossa, B. Rondeau, T. T. Pham and B. Fortin, Sci. Total Environ., 1998, 213, 193 CrossRef CAS.
- M. Horvat, S. Covelli, J. Faganeli, M. Logar, V. Mandic, R. Rajar, A. Sirca and D. Zagar, Sci. Total Environ., 1999, 238, 43 CrossRef.
- R. P. Mason, N. M. Lawson, A. L. Lawrence, J. J. Leaner, J. G. Lee and G. R. Sheu, Mar. Chem., 1999, 65, 77 CrossRef CAS.
- Y. H. Li, L. Burkhardt and H. Teraoka, Geochim. Cosmochim. Acta, 1984, 48, 1879 CrossRef CAS.
- A. Turner, G. E. Millward, A. J. Bale and A. W. Morris, Estuarine, Coastal Shelf Sci., 1993, 36, 1 Search PubMed.
- R. F. Anderson, P. H. Santschi, U. P. Nyffeler and S. L. Schiff, Can. J. Fish. Aquat. Sci., 1987, 44, 251 CAS.
- J. A. Campbell, R. Whitelaw, J. P. Riley, P. C. Head and P. D. Jones, Sci. Total Environ., 1988, 71, 141 CrossRef CAS.
- M. F. Fox, M. S. Johnson, R. T. Leah and D. Copplestone, Mar. Environ. Res., 1999, 47, 311 CrossRef CAS.
- G. W. Bryan and W. J. Langston, Environ. Pollut., 1992, 76, 89 CrossRef CAS.
- R. T. T. Rantala and D. H. Loring, Int. J. Environ. Anal. Chem., 1985, 19, 165 Search PubMed.
- L. L'Herroux, S. Le Roux, P. Appriou and J. Martinez, Environ. Pollut., 1997, 97, 119 CrossRef CAS.
- L. L'Herroux, S. Le Roux and P. Appriou, Mar. Pollut. Bull., 1998, 36, 56 CrossRef CAS.
- A. Turner and G. E. Millward, Estuarine, Coastal Shelf Sci., 1994, 39, 45 Search PubMed.
- J. Horowitz and K. A. Elrick, Appl. Geochem., 1987, 2, 437 CrossRef.
- P. Regnier and R. Wollast, Mar. Chem., 1993, 43, 3 CrossRef CAS.
- C. Gagnon, E. Pelletier, A. Mucci and W. F. Fitzgerald, Limnol. Oceanogr., 1996, 41, 428 Search PubMed.
- H. Hintelmann, P. M. Welbourn and D. R. Evans, Environ. Sci. Technol., 1997, 31, 489 CrossRef CAS.
- D. R. Turner, M. Whitfield and A. G. Dickson, Geochim. Cosmochim. Acta, 1981, 45, 855 CrossRef CAS.
- U. P. Nyffeler, Y. H. Li and P. H. Santschim, Geochim. Cosmochim. Acta, 1984, 48, 1513 CrossRef CAS.
- M. C. Stordal, P. H. Santschi and G. A. Gill, Environ. Sci. Technol., 1996, 30, 3335 CrossRef CAS.
- C. Gagnon and N. S. Fisher, Environ. Sci. Technol., 1997, 31, 993 CrossRef CAS.
- E. R. Sholkovitz, Earth Planet. Sci. Lett., 1978, 41, 77 CrossRef CAS.
- H. Biester and H. Zimmer, Environ. Sci. Technol., 1998, 32, 2755 CrossRef CAS.
- A. Turner and M. C. Rawling, Mar. Chem., 2000, 68, 203 CrossRef CAS.
- H. Bilinski, Z. Kwokal, M. Plavsic, M. Wrischer and M. Branica, Water Res., 2000, 34, 2001 CrossRef CAS.
- S. Watts, unpublished work..
- S. Meyer, G. Kubsch, M. Lovric and F. Scholz, Int. J. Environ. Anal. Chem., 1997, 68, 347 Search PubMed.
- W. Baeyens, C. Meuleman, B. Muhaya and M. Leemarkers, Hydrobiologia, 1998, 366, 63 Search PubMed.
- D. J. Mackey and A. Zirino, Anal. Chim. Acta, 1994, 284, 635 CrossRef.
- H. Bilinski, Z. Kwokal and M. Branica, Water Res., 1996, 30, 495 CrossRef CAS.
- J. A. Mastrine, J. C. J. Bonzongo and W. B. Lyons, Appl. Geochem., 1999, 14, 147 CrossRef CAS.
- D. A. Dzomback and F. M. M. Morel, in Surface Complexation Modeling. Hydrous Ferric Oxide, Wiley, New York, 1990. Search PubMed.
- R. Al-Farawati and C. M. G. van den Berg, Mar. Chem., 1999, 63, 331 CrossRef CAS.
- S. M. Theberge, G. W. Luther and A. M. Farrenkopf, Deep-Sea Res., Part II, 1997, 44, 1381 Search PubMed.
- P. D. Jones, PhD Thesis, University of Liverpool, Liverpool, 1978..
Footnote |
| † Presented at the Whistler 2000 Speciation Symposium, Whistler Resort, BC, Canada, June 25–July 1, 2000. |
| This journal is © The Royal Society of Chemistry 2001 |