Application of chemistry to in vitro diagnostic tests
Christopher P.
Price
Department of Clinical Biochemistry, St Bartholomew’s and the Royal London School of Medicine
& Dentistry, Turner Street, London, UK E1 2AD. E-mail: c.p.price@mds.qmw.ac.uk
First published on 28th November 2000
Abstract
Diagnostic testing for the screening, diagnosis and management of disease involves the detection and quantitation of a wide range of molecules, structures or whole cells in a complex biological matrix. Whilst separation techniques linked to physicochemical detection may be employed in the early investigation of a new marker, invariably the transition to routine application depends on the use of a biorecognition molecule, typically an enzyme or an antibody. In recent years the application of synthetic chemistry has led to the demonstration of synthetic biorecognition molecules—the search for specificity being aided by advances in combinatorial chemistry. Many of these sophisticated quantitative techniques are now encapsulated in prefabricated structures that facilitate automatic performance of the test to enable usage away from the laboratory, for example in the home or the workplace.
 Christopher P. Price Christopher P. Price | Christopher Price is Emeritus Professor of Clinical Biochemistry at the St Bartholomew’s and Royal London School of Medicine and Dentistry, and Director of Pathology for the Royal Hospitals NHS Trust. He trained as a clinical biochemist and has been involved in the development of a range of assays, including the use of novel bacterial enzymes for the quantitation of drugs and in light scattering immunoassay. He is also interested in point-of-care testing and has recently co-edited a book on the subject. |
1 Introduction
Diagnostic testing in the healthcare sector is primarily concerned with the recognition and confirmation of the presence of disease, supporting the management of that disease and assessing prognosis. It involves the detection (and often quantitation) of a vast range of analytes ranging from relatively simple species, such as ions, through complex organic molecules, such as drugs, hormones, and proteins to whole cells, such as a virus particle or plasma cell in a tissue section. Some examples of molecules of diagnostic interest are given in Table 1.| Ions | Potassium, calcium |
| Metabolites | Glucose, cholesterol |
| Drugs | Paracetamol, digoxin |
| Hormones | Cortisol, calcitriol |
| Proteins | Ferritin, albumin |
| Structures | Membranes, organelles |
| Cells | Bacterium, virus, plasma cell |
The analytical challenge that arises from the diversity of analytes of interest is compounded by the fact that the molecule of interest exists in a complex biological matrix, such as blood, plasma, sweat, urine, faeces or tissue biopsy. Thus interferences due to the existence of similar molecules, and to nonspecific effects, can be a real problem and must be investigated thoroughly and resolved before a method is set up for routine application.
It is important to recognise that the analytical performance characteristics of a particular test will influence both the clinical and operational effectiveness of the test. Thus, the analytical sensitivity of a method can determine whether the test meets the clinical requirement, e.g. in the case of the early detection of disease. The analytical sensitivity can also determine the amount of sample required and the incubation time—the latter being particularly important when rapid testing is required. Testing may be undertaken in a variety of situations varying from the home or the workplace, to the health centre, the ambulance or the emergency room, to the clinic or hospital ward. In many instances the needs of patients in these environments can be met by sending the sample to a clinical laboratory. However, there is an increasing proportion of testing that is undertaken at the point-of-care1 and by individuals who will not necessarily have received a technical training, including self testing. In this latter respect, albeit with the same parameters as at present, individuals are using diagnostic testing as an aid to the assessment of well being rather than to the recognition of disease; an example of this trend would be the measurement of cholesterol to support the use of dietary cholesterol lowering agents.
The analytical challenges are therefore associated with sensitivity (i.e. lowest detectable concentration) and specificity (i.e. accuracy for the analyte or group of analytes required), with speed of delivery and mobility, therefore size, and with simplicity of operation. The analytical sensitivity and specificity will have an important influence on the clinical sensitivity and specificity of the method, namely how good the test is at detecting people with and without the disease respectively. However, it should also be borne in mind that many tests, probably the majority, are not undertaken to determine whether the patient has a disease but rather to determine the severity of the disease and/or the effectiveness of the therapy.
2 Evolution of analytical methods
The early diagnostic tests were based on the primary senses of smell, sight and taste—with the apocryphal reference to physicians observing and tasting urine samples. In this respect the measurement of glucose provides an instructive case study, the use of taste being followed by simple chemical testing for reducing substances, as physicians, many with a good knowledge of chemistry, began to understand the characteristics of the disease, diabetes, and the properties of a key molecule, glucose. This was followed by the introduction of methods based on enzymatic catalysis and the use of the enzymes glucose oxidase, hexokinase or glucose dehydrogenase.2 The desire to achieve noninvasive monitoring of blood glucose, because of the clinical need for repeated measurements, has driven research workers to look at alternative spectroscopic techniques to obviate the trauma associated with repeated collections of blood samples.3All analytical techniques depend on the unique physicochemical properties of a target molecule, with the need for accurate quantitation in many cases. Physicochemical recognition can be based on techniques such as absorbance or fluorescence and on isoelectric point, molecular weight, size and shape and hydrophobicity being just a few examples. However, this relatively simple approach is rarely possible because of the complexity of the biological matrix of, for example, blood; it is therefore necessary to incorporate some form of separation method. Separation techniques that are used include electrophoresis, chromatography (for example, ion exchange or affinity), colloidal instability and solvent partition. A large proportion of clinically important biomarkers are measured in the plasma or serum fraction necessitating the removal of the cellular component of blood. Despite removal of the cellular components the serum or plasma still represents a complex matrix for many analytical methods, typical problems including the precipitation of proteins and turbidity associated with the presence of triglyceride containing particles.
However, whilst the use of separation techniques linked to detection technology based on physicochemical properties is satisfactory in the investigative stage of assay development, it becomes increasingly impractical as the workload increases or the assay is required away from a central laboratory facility. Consequently the developers of diagnostic devices have turned to harnessing biological phenomena—in essence mimicking the way in which organisms and higher species recognise molecules—in order to provide more practical analytical techniques for routine use.
3 Harnessing biology
All living organisms, of necessity, possess a variety of complex biochemical mechanisms that ensure the normal maintenance of life through the operation of metabolic cycles, the turnover of cells and tissue, of tissue repair as a consequence of trauma, protection against insult and changes in lifestyle. These mechanisms can be extremely specific and several have been exploited in the development of diagnostic tests (Table 2). The components of these biological systems provide the unique biorecognition for isolation of the molecule of interest within the complex matrix of the sample.| Enzymatic degradation |
| Antigen–antibody binding |
| Receptor–ligand binding |
| Oligonucleotide binding |
| Cell growth |
Enzyme mediated reactions
Reference has already been made to the measurement of glucose and the impact of enzyme catalysed reactions; these reaction sequences are not entirely unsurprising when one considers the role of glucose as an energy source for most living cells. There are many other examples where the enzymes from metabolic pathways have been isolated for use in the quantitation of metabolites.4An exciting development over the past two decades has been the isolation of enzymes from organisms that live in extreme environments, either in the presence of substances toxic to humans, or in extremes of temperature. The broad rationale adopted in the search for such organisms is based on adaptive mechanisms of the species that have enabled them to live on the molecule of interest as a source of carbon or nitrogen, together with survival in extreme environmental conditions, e.g. temperatures, salt concentration, etc. A typical example has been the isolation of a Pseudomonas species that metabolises the drug paracetamol; the metabolic pathway was shown to involve the removal of the acetate side chain releasing p-aminophenol which itself can be detected using a variety of chemical reactions or by electrochemical means. This discovery has led to the availability of a rapid method for detection and quantitation of the drug, that can be used throughout the day and night.5 Rapid access to a means of producing an accurate quantitative estimate of the drug level is an important requirement when attempting to recognise when a patient has taken an overdose of the drug and antidote must be given to avoid liver failure. In another example a detailed study of the properties of a phenylalanine dehydrogenase enabled the configuration of an assay that overcame the problems of cross reactivity with tyrosine so that a rapid method could be used for both screening and monitoring of patients with phenylketonuria.6 The availability of this method has replaced the need for cumbersome screening methods based on bacterial sensitivity and HPLC methods of quantitation.
There are many other examples where enzymes are used to provide rapid, accurate and precise measurements of a range of drugs and metabolites (Table 3).7 The unique specificity of an enzyme can also be complemented by stability with the use of enzymes obtained from thermophilic organisms. Protein chemists have also studied the differences between mesophilic and thermophilic organisms in order to determine what elements of the enzyme structures confer thermostability. Thus studies on the α-amylases (E.C. 3.2.1.1) derived from Bacillus licheniformis, stearothermophilus and amyloliquefaciens have shown that the enzyme of the first organism has 80 and 65% amino acid sequence homology with the other two enzyme sources respectively. The licheniformis enzyme also has a 100 fold greater half life at 90 °C than the amyloliquefaciens enzyme and, furthermore, is more stable at pH 11 than the latter.8
| Enzyme | Enzyme number | Analyte |
|---|---|---|
| Alcohol oxidase | EC 1.1.3.13 | Ethanol |
| Cholesterol oxidase | EC 1.1.3.6 | Cholesterol |
| Glucose dehydrogenase | EC 1.1.1.47 | Glucose |
| Uricase | EC 1.7.3.3 | Uric acid |
| Lactate dehydrogenase | EC 1.1.1.27 | Lactate |
| Salicylate monooxygenase | EC 1.14.13.1 | Salicylate |
| Glycerol dehydrogenase | EC 1.1.1.6 | Triglycerides |
| Aryl acylamidase | EC 3.5.1.13 | Paracetamol |
| Oxalate oxidase | EC 1.2.3.4 | Oxalate |
| Glutamate dehydrogenase | EC 1.4.1.3 | Ammonia |
| Urease | EC 3.5.1.5 | Urea |
| Phenylalanine dehydrogenase | EC 1.4.1.- | Phenylalanine |
Suzuki et al.9 constructed a variety of chaemeric genes from the structural genes of the licheniformis and amyloliquefaciens enzymes and studied their thermostability and irreversible inactivation. Their studies indicated that two regions of the central portion of the protein sequence were particularly associated with enhanced stability of the enzyme. They showed that replacement of alanine for lysine was one of the critical substitutions, an observation that has been made in the case of other thermophilic proteins. Of course there are other ways of enhancing enzyme stability, e.g. by protein crosslinking or covalent coupling to a solid phase. However, these approaches are inherently less desirable as they require additional manipulation of the enzyme preparation, which may in itself actually destroy the activity that one is seeking to protect.
Studies of the amino acid sequences and the folding of the protein structure have also been undertaken in order to determine the key factors that affect enzyme kinetics and substrate specificity. Thus in the case of lactate dehydrogenase, specific amino acid sequences have been identified for the binding of the nicotinamide coenzyme ring.10 Amino acids also define a mobile loop in the protein that moves when the substrate enters the active site, creating a substrate vacuole. The movement of the loop is the rate-limiting step and the amino acid sequence on the upper surface of the vacuole defines substrate specificity. It has been shown that removing a negative charge from the vacuole, with a positively charged amino acid inserted on the inside surface of the vacuole can modify the specificity of an enzyme from a lactate to a malate dehydrogenase. Thus, there is the potential to modify both the kinetics and specificity of an enzyme catalysed reaction as well as modifying the stability of the protein.
Antibody binding reactions
Whilst enzymes have been used successfully for the recognition of many small molecules, in other cases catalytic species have not been discovered. In the case of larger macromolecular species catalytic degradative reactions are of no value because invariably the product is not sufficiently distinctive to enable subsequent detection and quantitation. Consequently analytical chemists have turned to the immune response as a means of deriving a biorecognition molecule, to form the basis of an immunoassay.11 The generation of a polyclonal antiserum enabled the development, initially, of a competitive immunoassay and then a noncompetitive, so-called sandwich or immunometric assay. The format of these assays is illustrated in Fig. 1.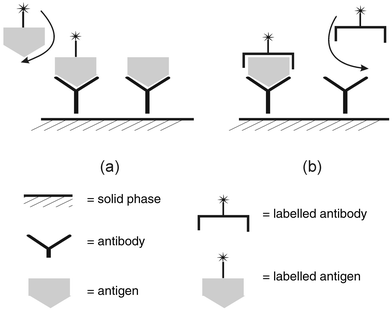 | ||
| Fig. 1 Outline of the format of (a) competitive and (b) noncompetitive (immunometric) heterogeneous immune assays. | ||
In the competitive format labelled antigen (a reagent) competes with sample antigen (target molecule) for a limited number of antibody binding sites coupled (as immunoglobulin) to a solid phase. After washing of the solid phase (e.g. tube, bead, magnetic particles) to remove unbound labelled antigen, the bound fraction is quantitated. The amount of label bound is inversely proportional to the amount of antigen in the sample. In the noncompetitive format sample antigen is captured by antibody coupled to the solid phase. The amount of antigen captured is then detected by the addition of labelled detection antibody. The amount of label coupled can be quantitated after the unbound fraction is removed and gives a signal in proportion to the sample antigen present.
Since these analytical concepts were developed in the 1960s three significant developments have influenced the evolution of immunoassay technology. In 1975 Köhler and Milstein described the production of monoclonal antibodies.12 In the context of diagnostic testing this enabled the isolation of antibodies that recognised unique epitopes on a molecule (as against the situation with a polyclonal antiserum which contains a population of antibodies which might recognise a range of epitopes). The use of a dissimilar pair of monoclonal antibodies (i.e. recognising different and distinct epitopes) in an immunometric assay meant that it was possible to isolate a defined molecule in a mixture that contained fragments or polymeric aggregates. Thus, as an example, it became possible to quantitate the whole parathyroid hormone molecule in the presence of fragments, providing a more accurate correlation with disease activity.13 Several tumour markers were also discovered, and their measurement applied in clinical practice, as a consequence of the application of monoclonal antibody technology.14
It became evident that the use of radioisotopic labels was undesirable due to the safety limitations on their use and also because of constraints on assay sensitivity (detection limits). A range of alternative labels have subsequently been employed with concomitant reduction in detection limits (see Fig. 2) as well as less restriction on utilisation—the main demonstration of this being the development of automated immunoassay analysers.15,16 In this case the use of an enzyme was not designed to take advantage of its specificity but rather of its (hopefully) high turnover number leading to the possibility of signal amplification.
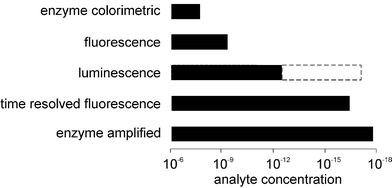 | ||
| Fig. 2 A summary of some nonisotopic labels employed in heterogeneous immunoassays and the analytical ranges over which they can be employed. | ||
The advent of nonisotopic labels subsequently led to the development of strategies for modulation of the properties of the label in the presence of the binding partner. The first commercial application of this approach was the enzyme multiplied immunoassay (EMIT) in which the activity of an enzyme such as glucose-6-phosphate dehydrogenase coupled to a hapten was inhibited on binding of antibody to the labelled hapten. (A hapten is a small molecule which in itself is not capable of eliciting an immune response, i.e. generating antibodies when injected into an animal, but which can generate specific antibodies when it is coupled to a carrier protein.) Consequently there was no necessity to undertake a separation of free from bound label, binding being differentiated by modulation of activity, and the homogeneous immunoassay was born. Subsequently, other modulation techniques have been described and several commercialised; these include fluorescence polarisation, fluorescence excitation transfer, substrate labelled, inhibitor labelled and cloned enzyme donor immunoassays.17 Some alternative approaches to homogeneous immunoassays have not depended on modulation of a signal from a label, but rather depended on properties associated with binding of antigen to antibody, e.g. light scattering, surface plasmon resonance.18,19
4 Mimicking biology
Reference has already been made to achieving a greater understanding of the molecular chemistry of enzyme specificity and kinetics, as well as the stabilisation of proteins. Considerable work has also gone into enhancing understanding of the physicochemical characteristics of the antigen–antibody reaction. This has been of considerable benefit in optimising immunoassay procedures as well as in the construction of better antibodies or antibody mimics.The paratope (binding) region of the antibody molecule is thought to be represented by discontinuous sequences of amino acids, specificity being conferred by the unique sequences in the three complementarity determining regions (CDRs) of the light and heavy chains, as well as their spatial configuration. It is thus a composite of primary and second binding reactions and overcoming of the inherent repulsion between protein molecules that defines the kinetics and specificity of the antigen–antibody binding reaction. The interaction between residues in the epitope and paratope regions include: (i) hydrophilic repulsion operating over a distance of 20–30 Å; (ii) primary attraction due to hydrophobic and electrostatic interactions operating over distances of 30–100 Å; and (iii) secondary interaction due to van der Waals and hydrogen bonding operating over 2–10 Å. Modification of the ionisation status of the amino acid and residues, together with changes in the water layer that surrounds the protein molecules can radically alter the influence of the competing repulsive and attractive forces, thereby altering the rate of reaction as well as the risk of nonspecific binding. Changes in the amino acid sequences, particularly associated with the CDRs can influence the specificity of binding.20
There are several limitations to the use of antibodies produced by conventional means, including the need to use an animal, animals not tolerating all immunogen molecules, the process of antibody production—particularly monoclonals—being laborious, antibodies being sensitive to denaturation and reaction kinetics which are not always favourable. Some of the approaches to overcoming these problems—broadly termed antibody engineering—include displaying peptide libraries on phages21 and ribosomes,22 modified paratope sequences23 and in vitro immunisation.24
The production of an antibody with the desired specificity and binding characteristics depends to a certain extent on chance, and the odds of isolating an antibody with the required characteristics lengthen when the analyte differs by only a small degree from other molecular species, e.g. some steroid hormones, glycated versus nonglycated proteins. Attempts to improve specificity have employed the use of synthetic immunogens and of synthetic antibodies. Two examples illustrate the challenges associated with the differentiation between similar but distinctly individual antigens. In an attempt to produce an immunoassay for the quantitation of the bone isoform of alkaline phosphatase Hill and Wolfert25 screened a total of 20441 monoclonal antibody clones to achieve less than 3 percent crossreactivity toward the liver isoform. The two isoforms differ only in the post translation sialylation of the same gene product. The commercial product that resulted from this development in fact showed a crossreactivity of 7–12% toward the liver isoform (depending on the method of calculations)26 presumably reflecting a compromise on performance to achieve a commercial product. On the other hand a successful approach taken to achieving an antibody that would discriminate between glycated and nonglycated haemoglobin was to raise a monoclonal antibody against an artificial immunogen based on a glycated peptide mimicking the glycated N-terminal sequence of the globin molecule.27
One of the early approaches to the production of antibody mimics has been the construction of an antigen binding pocket in a polymer. The core of the technology lies in the polymerisation of monomers in the presence of the target molecule. The monomers employed are capable of some form of bonding with specific functional groups on the target molecules—the key being that they are reversible interactions. The types of bonding are those that take place in antigen–antibody interactions, namely electrostatic bonding and hydrophobic interactions. The polymerisation process effectively fixes the interacting monomers in space around the target molecule. Elution of the target molecule leaves a binding pocket fixed in the polymer. Several assays for small molecules have been demonstrated.28,29 Alternative approaches employing the basics of combinatorial chemistry have also been demonstrated with sequences of nucleotides or of amino acids.30,31 The random oligonucleotide sequence capable of binding to an analyte of interest is known as an aptamer. Initially a random sequence library of oligonucleotides is produced by combinatorial chemical synthesis of DNA. Each of the oligonucleotides is a linear sequence; a library containing a 40-nucleotide random region is capable of generating 1024 individual sequences. In practice, the synthesis produces between 1014 and 1015 sequences but it is still claimed that the molecular diversity exceeds that achieved with other techniques to date, such as peptide libraries used for phage display.31
Screening of oligonucleotide libraries is usually undertaken with an immobilised form of the target molecule. After removal of unbound sequences the bound oligonucleotide can be collected and amplified to obtain an enriched library. By varying the binding conditions (for stringency) the sequences with greatest affinity for the target molecule can be selected. To date, aptamers with unique specificities have been demonstrated, being able to discriminate small changes in structure, e.g. the Dvs.L enantiomer of the target molecule.32
There is a more limited literature on the use of combinatorial approaches to the generation of peptide sequences. Combinatorial linear peptides have been shown to bind to large target molecules,33 although a question remains over the affinity and specificity that can be achieved. It is thought that this may be due to the limited stability that can be achieved with linear peptides. There are more examples of linear peptides bound to small target molecules, e.g. amino acids.34
The strategy adopted in the search for binding peptide sequences is firstly to generate a library of random sequences coupled individually to a solid phase, e.g. a bead or magnetic particle. Following the search for selective binding and isolation, the sequence can be identified using mass spectrometry. Further iterations of a more restricted sequence of amino acids obtained from the initial analysis can then be screened under more stringent conditions, e.g. a shorter incubation time to isolate the higher affinity binding sequences. The techniques are not limited to the recognition of small molecules and molecular imprints of whole organisms have been described.35 Recently the creation of a synthetic binding pocket has been extended to the creation of a biorecognition surface,36 which may have interesting applications in the field of biosensors.
One of the perceived limitations of the antigen–antibody reaction, compared with an enzyme mediated reaction, has been the relative irreversibility of the former. This has meant that it has been difficult to develop immunoassay based sensors for continuous monitoring. This problem has been overcome in some of the optical immunosensors by appropriate choice of reaction conditions.19 However, more recently there have been exciting developments described for the use of reversibly antigen responsive hydrogels,37 which open up some interesting possibilities for the future.
5 In search of faster reactions
Time is an important resource from a clinical standpoint and there is a continuing demand for faster delivery of results, be it due to an emergency, the time spent in a clinic or the impatience of the user. More research is therefore now being undertaken to shorten the “time to result”, there being many clearly proven examples of faster treatment leading to better patient outcomes, e.g. the use of thrombolytic agents in patients who have suffered a myocardial infarction. One easy way to achieve this in enzyme and antibody mediated reactions is to perform a kinetic assay, i.e. not run a reaction to end point or equilibrium. However, this can impose other constraints on the assay system. Perhaps the obvious way to increase the reaction rate is to increase the incubation temperature; this however can only be employed to a limited degree because of the lability of the reagents (enzymes or antibodies) and of constituents of the sample.One approach to improving reaction kinetics in immunoassay is to employ the addition of polyethylene glycol. It is thought that the polymer removes the water shell around the protein, reducing the repulsive forces and thereby promoting binding. However, removal of the water shell around proteins reduces their colloidal stability and may lead to agglutination. In fact, the use of such glycols is one of the accepted techniques for fractional protein precipitation.
One exciting approach that has been reported for immunoassay is the use of ultrasound. Using latex particle labelled antibodies Gray et al.38 demonstrated a significant reduction in reaction time with improved detection limits. The principle of the technique is based on the fact that latex particles in an ultrasonic standing wavefield are driven toward pressure nodal regions. Consequently the concentration of particles in these regions is markedly increased, which enhances the rate of collision between particles and the antigen present, thereby increasing the rate of agglutination, as well as lowering the detection limit. Using an image analyser to detect and quantitate the agglutination Thomas and Coakley39 demonstrated a detection limit of 230 ng L−1 in an assay for C reactive protein. Another approach that has also been exploited is the use of alternating current.40 However, these opportunities remain largely unexplored at present although there are considerable gains to be made by improving reaction rates in terms of both the productivity of automated analysers and also the practical application of point-of-care testing devices (shorter time to result).
6 Prefabricated analytical devices
The realisation that certain diagnostic tests were required with an urgency or convenience that precluded dispatch of a sample to a central laboratory has led to the construction of prefabricated devices that achieved several objectives: (i) safe storage of reagents prior to use; (ii) performance of complex chemical reactions on the simple addition of sample; (iii) rapid production of a result; and (iv) safe containment of samples and reagents for disposal. The analytical steps encapsulated in a typical device for an immunoassay are indicated in Table 4.| Sample metering |
| Plasma separation |
| Reagent metering |
| Reactant mixing |
| Incubation period |
| Free/bound separation |
| Signal detection |
| Procedural control |
| Result calculation |
| Result reporting |
| Reactant disposal |
There are three basic types of device that employ: (i) some form of reaction cuvette; (ii) a porous matrix; or (iii) an optical surface.41 The dimensions of the devices vary considerably, but in order to reduce reactant volumes (and thus reaction times), and enable greater mobility, there is a considerable drive to miniaturise such devices.
In a reaction cell type of device the reagents are either lyophilised and separated from a diluent, or maintained in stabilised solution. There is some form of sample addition port and fluidics circuit to bring the reactants together; in one example the movement of fluids was achieved by centrifugation.42 In other examples the mixing is achieved by releasing a barrier and the use of magnetic ball bearings. The optical density is read through an optical window in the cuvette.
The most common form of prefabricated device is the so-called ‘stick test’ which comprises one of two types: (i) simple reagent pads;43 or (ii) liquidic circuits. In their simplest form the sample added to one end of a paper strip migrates along the strip passing across reagent impregnated regions, capturing antibody in the case of an immunoassay.41 It is thus possible to perform a heterogeneous immunoassay, which can be quantitated if the colour captured can be read by reflectance (see Fig. 3). Many of these devices are qualitative or semi-quantitative and yet they can be optimised to give extremely accurate discrimination (cut off values). This has been illustrated in an example of such a device for testing for drugs of abuse.44
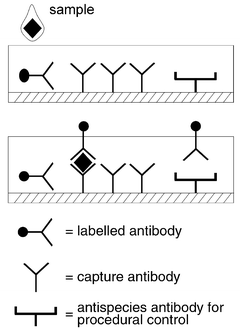 | ||
| Fig. 3 Schematic diagram of a porous matrix immunoassay device. | ||
The third type of device is based on the bioreactive surface which is linked to a means of signal transduction—the biosensor. A whole range of biosensing principles have been described and are too numerous to set out in detail. However, the approaches that have been successful are based on electrochemical, optical diffraction and evanescent wave technologies. The electrochemical detection has been successful in the case of the glucose sensor.45 Surface plasmon resonance has, to date, been one of the more successful technologies employed in immunosensors.19,46
Whilst the cuvette style devices are very dependent on accurate machining of the fluidics circuits, the porous matrix and surface based systems depend more heavily on optimisation of the surface chemistry, in addition to the chemical reactions that support the biorecognition system. In all three approaches, however, the success of the prefabricated devices is totally dependent on the reproducibility of manufacture.
7 Conclusion
Whilst many of the routine diagnostic tests now employed in the diagnosis and management of disease are based on biorecognition molecules, such as enzymes and antibodies, our understanding of these complex chemical reactions has led to technological developments that have improved both application and performance. Thus, in the future we are likely to rely less on biotechnological innovation to produce new enzymes and antibodies, looking rather to the chemical synthesis of novel recognition molecules with the desired specificity and reaction kinetics.This review has not dealt with the field of molecular diagnostics, that is the probing of DNA for the recognition of variations in human DNA, for the diagnosis of inherited disorders or for the recognition of predisposition diseases which have a genetic element in their aetiology. This really constitutes a review in its own right and has been covered in many reviews and books as well as also being a topic covered in the meeting from which this article was written. It is, however, worth speculating on the genomic revolution and its impact on the development and utilisation of new diagnostic tests.
There are now many who realise that the work of the genome project has only just begun. Thus it is the information that derives from knowledge of the human genome sequence that will increase our understanding of disease processes.47,48 It is then that new markers will be developed that can be used to support the diagnosis and management of disease. Thus, whilst many might disagree with this prediction, there is little evidence to date to indicate that molecular diagnostic tests will predominate in the laboratories of the future in relation to human disease. On the other hand, there is no doubt that molecular recognition will become the preferred approach to microbiological and virological diagnosis.
8 References
- Point-of-Care Testing,eds. C. P. Price and J. M. Hicks, AACC Press, Washington, 1999, pp. 580. Search PubMed.
- Measurement of blood glucose, J. M. Burrin and C. P. Price, Ann. Clin. Biochem., 1985, 22, 327 Search PubMed.
- Spectroscopic and clinical aspects of noninvasive glucose measurements, O. S. Khalil, Clin. Chem., 1999, 45, 165 Search PubMed.
- Methods of Enzymatic Analysis, eds, H. U. Bergmeyer, J. Bergmeyer and M. Graßl, 3rd edn., VCH Verlagsgesellschaft, Germany, 1986, (12 volumes). Search PubMed.
- Development of an enzyme-based assay for acetaminophen, P. M. Hammond, M. D. Scawen, T. Atkinson, R. S. Campbell and C. P. Price, Anal. Biochem., 1984, 143, 152 CrossRef CAS.
- Development and validation of a robust specific enzyme mediated assay for phenylalamine in serum R. S. Campbell, G. M. Brearley, H. Varsani, H. C. Morris, T. P. Milligan, S. K. Hall, P. M. Hammond and C. P. Price, Clin. Chim. Acta, 1992, 210, 197 CrossRef CAS.
- Novel enzymes as reagents C. P. Price, R. S. Campbell and P. M. Hammond, Clin. Chim. Acta, 1995, 237, 3 CrossRef CAS.
- Why is one Bacillus α-amylase more resistant against irreversible thermoinactivation than another? S. J. Tomazic and A. M. Klibanov, J. Biol. Chem., 1998, 263, 3092.
- Amino acid residues stabilizing a Bacillus α-amylase against irreversible thermoinactivation, Y. Suzuki, N. Ito, T. Yuuki, H. Yamagata and S. Udaka, J. Biol., 1989, 264, 18933 Search PubMed.
- Design and synthesis of new enzymes based on the lactate dehydrogenase framework, C. R. Dunn, H. M. Wilks, D. J. Halsall, T. Atkinson, A. R. Clarke, H. Muirhead and J. J. Holbrook, Philos. Trans. R. Soc. London, Ser. B, 1991, 332, 177 Search PubMed.
- C. L. Knott, K. Kuus-Reichel, R.-S. Liu and R. L. Wolfert, Development of antibodies for diagnostic assays, in Principles and Practice of Immunoassay, eds. C. P. Price and D. J. Newman, 2nd edn., Macmillan Reference Ltd, London, 1997, pp. 37–64. Search PubMed.
- Continuous cultures of fused cells secreting antibody of predefined specificity, G. Köhler and C. Milstein, Nature, 1975, 256, 495.
- Highly sensitive two-site immunoradiometric assay of parathyrin, and its clinical utility in evaluating patients with hypercalcemia, S. R. Nussbaum, R. J. Zahradnik, J. R. Lavigne, G. L. Brennan, K. Nozawa-Ung, L. Y. Kim, H. T. Keutmann, C. A. Wang, J. T. Potts, Jr. and G. V. Segre, Clin. Chem., 1987, 33, 1364 Search PubMed.
- Radioimmunometric assay for a monoclonal antibody-defined tumor marker CA 19-9, B. C. Del Villano, S. Brennan, P. Brock, C. Bucher, V. Liu, M. McClure, B. Rake, S. Space, B. Westrick, H. Schoemaker and V. R. Zurawski, Jr., Clin. Chem., 1983, 29, 549 Search PubMed.
- Selected strategies for improving sensitivity and reliability of immunoassays L. J. Kricka, Clin. Chem., 1994, 40, 347 Search PubMed.
- D. W. Chan, Automation of immunoassays, in Immunoassay, eds. E. P. Diamandis and T. K. Christopoulos, Academic Press, San Diego, 1996, pp. 483. Search PubMed.
- J. P. Gosling, Enzyme immunoassay: with and without separation, in Principles and Practice of Immunoassay, eds. C. P. Price and D. J. Newman, 2nd edn., Macmillan Reference Ltd, London, 1997, pp. 351–88. Search PubMed.
- C. P. Price and D. J. Newman, Light-scattering immunoassay, in Principles and Practice of Immunoassay, eds. C. P. Price and D. J. Newman, 2nd edn., Macmillan Reference Ltd, London, 1997, pp. 445–80. Search PubMed.
- Immunosensors: technology and opportunities in laboratory medicine C. L. Morgan, D. J. Newman and C. P. Price, Clin. Chem., 1996, 42, 193 Search PubMed.
- VH shuffling can be used to convert an Fv fragment of anti-hen egg lysozyme specificity to one that recognises a T-cell receptor V alpha E. S. Ward, Mol. Immunol., 1995, 32, 147 CrossRef CAS.
- Peptides on phage: a vast library of peptides for identifying ligands S. E. Cwirla, E. A. Peters, R. W. Barrett and W. J. Dower, Proct. Natl. Acad. Sci. USA, 1990, 87, 6378 Search PubMed.
- Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries J. Hanes, L. Jermutus, S. Weber-Bornhauser, J. R. Bosshard and A. Pluckthun, Proc. Natl. Acad Sci. U.S.A., 1998, 95, 14130 CrossRef CAS.
- Towards the design of an antibody that recognizes a given protein epitope P. M. Kirkham, D. Neri and G. Winter, J. Mol. Biol., 1999, 285, 909 CrossRef CAS.
- Immunization in vitro and production of monoclonal antibodies specific to insoluble and weakly immunogenic proteins J. Van Ness, U. K. Laemmli and D. E. Pettijohn, Proc. Natl. Acad. Sci. U.S.A., 1984, 81, 7897 CAS.
- The preparation of monoclonal antibodies which react preferentially with human bone alkaline phosphatase and not liver alkaline phosphatase C. S. Hill and R. L. Wolfert, Clin. Chim. Acta, 1989, 186, 315 CrossRef.
- Direct comparison of performance characteristics of two immunoassays for bone isoform of alkaline phosphatase in serum C. P. Price, T. P. Milligan and C. Darte, Clin. Chem., 1997, 43, 2052 Search PubMed.
- Adaptation of latex-enhanced assay for percent glycohemoglobin to a Dade Dimension® analyzer P. Holownia, E. Bishop, D. J. Newman, W. G. John and C. P. Price, Clin. Chem., 1997, 43, 76 Search PubMed.
- The emerging technique of molecular imprinting and its future impact on technology K. Mosbach and O. Ramström, Biotechnology, 1996, 14, 163 Search PubMed.
- Synthesis of molecular imprinted polymer networks S. Rimmer, Chromatog., 1998, 47, 470 CAS.
- Aptamers as therapeutic and diagnostic reagents: problems and prospects S. E. Osborne, I. Matsumura and A. D. Ellington, Curr. Opin. Chem. Biol., 1997, 1, 5 CrossRef CAS.
- Aptamers: an emerging class of molecules that rival antibodies in diagnostics S. D. Jayasena, Clin. Chem., 1999, 45, 1628 Search PubMed.
- RNA aptamers that bind L-arginine with sub-micromolar dissociation constants and high enantioselectivity A. Geiger, P. Burgstaller, H. von der Eltz, A. Roeder and M. Famulok, Nucleic Acids Res., 1996, 24, 1029 CrossRef CAS.
- In vitro diagnostics in diabetes: meeting the challenge A. P. F. Turner, B. Chen and S. A. Piletsky, Clin. Chem., 1999, 45, 1596 Search PubMed.
- Investigation of the molecular recognition of amino acids by cyclopeptides with reflectometric interference spectroscopy D. Liepert, D. Nopper, M. Bauser, G. Gauglitz and G. Jung, Angew. Chem. Int. Ed., 1998, 37, 3308 CrossRef.
- Bacteria-mediated lithography of polymer surfaces A. Aherne, C. Alexander, M. J. Payne, N. Perez and E. N. Vulfson, J. Am. Chem. Soc., 1996, 36, 8771 CrossRef CAS.
- Template-imprinted nanostructured surfaces for protein recognition H. Shi, W.-B. Tsai, M. D. Garrison, S. Ferrari and B. D. Ratner, Nature, 1999, 398, 593 CrossRef CAS.
- A reversibly antigen-responsive hydrogel T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766 CrossRef CAS.
- Ultrasound-enhanced latex immunoagglutination and PCR as complementary methods for non-culture-based confirmation of meningococcal disease S. J. Gray, M. A. Sobanski, E. B. Kacmarski, M. Guiver, W. J. Marsh, R. Borrow, R. A. Barnes and W. T. Coakley, J. Clin. Microbiol., 1999, 37, 1797 CAS.
- Measurement of antigen concentration by an ultrasound-enhanced latex immunoagglutination assay N. E. Thomas and W. T. Coakley, Ultrasound in Med. & Biol., 1996, 22, 1277 CrossRef CAS.
- Multisample analysis using an array of microreactors for an alternating-current field-enhanced latex immunoassay M. I. Song, K. Iwata, M. Yamada, K. Yokoyama, T. Takeuchi, E. Tamiya and I. Karube, Anal. Chem., 1994, 66, 778 CrossRef CAS.
- C. P. Price and G. Thorpe, Disposable analytical devices in point-of-care testing, in Point-of-Care Testing, eds. C. P. Price and J. M. Hicks, AACC Press, Washington, 1999, pp. 17–40. Search PubMed.
- Two-dimensional centrifugation for desktop clinical chemistry S. G. Schultz, J. T. Holen, J. P. Donohue and T. A. Francoeur, Clin. Chem., 1985, 31, 1457 Search PubMed.
- Performance of a reagent strip device for quantitation of the urine albumin:creatinine ratio in a point-of-care setting M. Parsons, D. J. Newman, M. Pugia, R. G. Newall and C. P. Price, Clin. Nephrol., 1999, 51, 220 Search PubMed.
- Simultaneous detection of seven drugs of abuse by the Triage™ panel for drugs of abuse K. F. Buechler, S. Moi, B. Noar, D. Mcgrath, J. Villela, M. Clancy, A. Shenhav, A. Collegmore, G. Valkirs, T. Lee, J. F. Bruni, M. Walsh, R. Hoffman, F. Ahmuty, M. Nowakowski, J. Buechler, M. Mitchell, D. Boyd, N. Stiso and R. Anderson, Clin. Chem., 1992, 38, 1678 Search PubMed.
- Recent advances in amperometric glucose biosensors for in vivo monitoring S. A. Jaffari and A. P. F. Turner, Physiol. Med., 1995, 16, 1 Search PubMed.
- D. R. Purvis, D. Pollard-Knight, C. R. Lowe, Direct immunosensors, in Principles and Practice of Immunoassay, eds. C. P. Price, D. J. Newman, 2nd edn., Macmillan Reference Ltd, London, 1997, pp. 513–43. Search PubMed.
- Protein function in the post-genomic era D. Eisenberg, E. M. Marcotte, I. Xenarios and T. O. Yeates, Nature, 2000, 405, 823 CrossRef CAS.
- Proteomics to study genes and genomes A. Pandey and M. Mann, Nature, 2000, 405, 837 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |