Getting into shape: Conformational and supramolecular landscapes in small biomolecules and their hydrated clusters
Evan G. Robertson† and John P. Simons
Physical and Theoretical Chemistry Laboratory, South Parks Road, Oxford, UK OX1 3QZ
First published on 13th December 2000
Abstract
The last few years have seen a very rapid growth in understanding the influence of non-bonded, particularly hydrogen-bonded interactions, on the shapes and conformations of flexible molecules, including those of pharmacological or biological importance, and of the supramolecular structures of their hydrated clusters. This has come about through the combination of a wide range of newly developed spectroscopic strategies, many of which are laser-based, coupled with powerful and widely available ab initio codes for structural computation. The consequent rapid growth of a new link between the worlds of chemistry and biophysics is surveyed in a review which introduces the range of present strategies, their origins, and their application to studies of neutrotransmitters, amides and peptides, amino acids and nucleic acid bases. It concludes with a prospectus for the future.
1. A co-operative interaction
Molecular shape, molecular conformation and molecular charge distribution play crucial roles in the selectivity and function of biologically active molecules. Molecular shape and conformation are controlled by a delicate balance between the forces operating through covalent bonds, i.e. those which determine their ‘skeletal’ structures, and the so-called ‘non-bonded interactions’, operating through space between neighbouring groups of atoms or local charges within the molecules, or between the molecules and their local environment. Together, these determine their conformational landscapes and preferred architectures. Molecular shape, and the interactive forces between the molecule and its neighbourhood, also play a crucial role in the molecular transport and molecular recognition processes that are involved in almost all aspects of biological function, ranging from transport through membranes, to neurotransmission, to specific drug-receptor interactions, to enzyme catalysis. The ubiquitous hydrogen-bonded interaction, operating both within the molecules and externally with neighbouring molecules, especially water (Nature’s favoured solvent, upon which life depends), are particularly important. The subtle balance between intra- and inter-molecular hydrogen-bonded interactions in controlling biomolecular conformations, the delicate balance between hydrogen-bonded and other ‘non-bonded’ interactions, and the way in which co-operative interactions may control the specificity of molecular function, remain very unclear.In the last few years, using a powerful combination of molecular electronic and/or vibrational spectroscopy, or microwave spectroscopy, coupled with ab initio computation, it has become possible to begin to address directly, some of these fundamental biophysical questions. The ‘structural images’ of spectrally resolved biomolecular conformers and their mass-selected, often hydrogen-bonded, molecular clusters, isolated in the gas phase and/or frozen in a low temperature environment, have been encoded in their rotationally and/or vibrationally resolved spectra. Conversion of the images into structural assignments has depended crucially on the support provided by high-level ab initio quantum chemical calculation: indeed, it is the synergism between experiment and ab initio computation that has made the enterprise possible. Theory and experiment enjoy a symbiotic relationship—their interaction is a co-operative one. Theory provides the ‘ á la carte menu’ of structural possibilities; experimental observation and analysis tells us which ones are actually chosen. The strategy borrowed from the world of chemical physics, although reductionist, recognises that complexity grows out of simplicity.
Primary objectives include:
(i) Characterisation of the conformational and structural landscapes of small, pharmacologically important molecules, biomolecular building blocks and small biomolecules, and their supramolecular hydrated or clustered assemblies, isolated in the gas phase.
(ii) Characterisation of the balance of molecular interactions which govern their structures, particularly in hydrated environments.
(iii) Characterisation of ligand–receptor interactions at model binding sites, a vital first step in understanding the mechanisms and consequences of molecular recognition.
Target systems have included neurotransmitters and hormones; amino acids, amides and small peptides; model and real nucleic acid bases; enzyme mimics and inhibitors; and their size-selected clusters and molecular hydrates. The aspiration is a quantitative understanding of the interplay between molecular interaction and molecular shape, and ultimately, the influences these have on molecular reactivity in model biochemical systems. There is a long way to go. This article presents some snapshots of current laser-based experimental strategies for probing the structure of small biomolecular systems, and the exciting new results they are generating; it also provides some route maps leading to possible future developments. While acknowledging the great debt owed to early pioneers and present travellers along the way, the snapshots will be taken largely through the authors' own, admittedly narrow aperture lens.
2. Strategies
Jet cooling
Spectroscopic measurements of molecules isolated in the gas phase or better, entrained in the low temperature environment of a seeded, free jet expansion1–4 provide a very powerful experimental strategy for the isolation and detection of small, biologically important molecular conformers (or isomers or tautomers) as well as their intermolecular clusters. Involatile systems, e.g. di- and tri-peptides incorporating tryptophan and glycine, can be ‘encouraged’ to evaporate into the expanding gas stream, by using laser ablation and/or ‘thermospray’ techniques.5–8 If the potential energy barriers separating individual conformers are sufficiently high, their interconversion is suppressed. As a ‘rule of thumb’, barriers greater than ca. 12–15 kJ mol−1 appear sufficient to ensure that the Boltzmann population distributions maintained among conformers prior to the expansion stage, will be preserved during supersonic cooling.9,10 Comparisons of experimental population distributions (estimated from relative spectral intensities) with those based upon high-level ab initio calculation, do not always agree, however.11 Some conformations appear relatively over-populated, or under-populated, or are absent altogether.11,12 Assuming their absence is not due simply to low spectral intensity, and the quantitative reliability of high-level ab initio predictions of their relative energies (or free energies at the pre-expansion temperature) this behaviour signals the incidence of collisional relaxation during the free jet expansion phase.11,12 Kinetic control begins to become important when there are low energy pathways connecting the alternative conformational potential energy wells. Again, as a ‘rule of thumb’, (zero point) energy barriers lower than ca. 5–6 kJ mol−1 appear to be small enough to allow collisional relaxation while the gas is still cooling, and a consequent draining of population into the conformational structures of lower zero point energy.11,12Kinetic and thermodynamic factors also combine to control the relative populations of molecular clusters, since these are formed during the free jet expansion. Their size distribution will move towards higher stoichiometries with increased cooling of the seeded gas expansion and their relative populations will depend upon the selected expansion conditions (e.g. the nozzle aperture dimensions, the stagnation pressure, the number densities and the laser probe delay-time, in a pulsed jet experiment). In addition, although size-selected clusters of a given molecular conformer may have a considerable number of alternative structures, it is only the lowest lying one or two that are generally detected.
Detection and identification
Molecular electronic spectra, recorded through the excitation of their LIF and/or mass-selected, resonant two photon ionisation (MS-R2PI), often present a tangle of overlapping spectral features, associated with the large family of clusters, conformers or isomers that can be created and/or stabilised within any given system.4 If the R2PI signals are mass selected, the carriers of the individual spectra can be identified. In the earliest measurements, the contributions made by individual conformers were distinguished from bands associated with vibronic excitation, by recording the change (or rather, the absence of any change) in their relative intensities when the laser power was sufficient to saturate their optical transitions.13 Nowadays, double resonance ‘hole-burn’ strategies provide a much more powerful approach allowing all contributions, whether from distinct conformers, isomers, individual clusters, or even impurities, to be separately identified.14 In the UV–UV scheme shown in Fig. 1(a), UV radiation from the probe laser is selectively tuned to the spectral feature of interest (A, B or C), while an intense UV ‘burn ’ laser is scanned across the spectrum to promote selective ‘hole-burning’ by transferring population out of the probed level. Vibronic features associated with excitation out of the selected level, are revealed by the ‘dips’ in their associated LIF or R2PI signals. In the alternative IR–UV scheme15–17 shown in Fig. 1(b), an IR ‘burn’ laser is tuned into resonance with a vibrational mode of a single species only, while the UV probe laser is scanned. In this strategy, an UV spectrum is obtained in which the feature(s) associated with the selected species is (are) greatly reduced or even totally removed.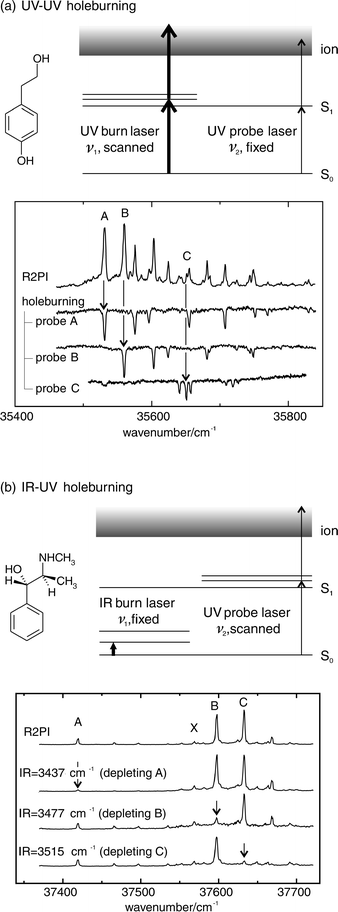 | ||
| Fig. 1 (a) UV–UV and (b) IR–UV hole-burning schemes, illustrated by the examples of tyrosol and pseudoephedrine. | ||
Double resonance excitation offers many other advantages:
for example, if tuneable, but low intensity narrow-line laser excitation
is used to excite the initial transition into the intermediate
molecular electronic state (followed by its subsequent i
onisation using a second high intensity laser), the MS-R2PI spectrum
can be recorded under conditions that avoid saturation, thereby retaining the integrity of the rotational band contours. Tuneable, two-colour excitation also allows the MS-R2PI spectra of molecular conformers or clusters to be recorded
close to threshold, where the ‘soft ionisation
’ conditions
minimise the degree of cluster ion fragmentation. Finally,
making a virtue out of necessity, tuneable two-colour excitation
can be used to promote, rather than minimise cluster
ion dissociation. Measurements of the dissociation and molecular
ionisation thresholds then provide a means of determining
the binding energies of size-selected molecular clusters in
the ground electronic state,18 see Fig. 2. Alternatively, cluster
binding energies can be determined through measurements of
their vibrational predissociation thresholds, either in the intermediate
electronically excited state,19 or in the ground state
(accessed ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia stimulated emission pumping).20
ia stimulated emission pumping).20
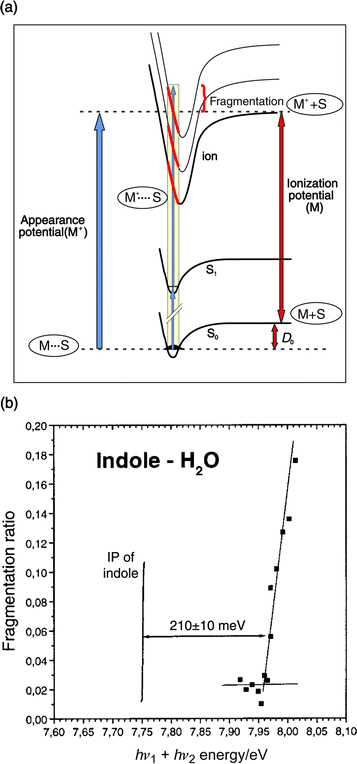 | ||
| Fig. 2 (a) Scheme for determination of ground state binding energies, by dissociative resonant two-photon ionisation. (b) Fragmentation efficiencies obtained after 2-colour ionisation of the indole–H2O complex, as a function of total photon energy hν1 + hν2. | ||
Of course these are not the only spectroscopic approaches
that have led the way in probing the structures of small bio
molecular systems. As far back as the late 1970s Suenram and
his co-workers21 and the group at Monash led by Brown,22 pioneered
the use of microwave, and millimetre wave spectroscopy,
coupled with ab initio
computation, to characterise some
of the alternative conformational structures of a range of aliphatic
amino acids. The Monash group also used high resolution
pure rotational spectroscopy to infer the tautomeric (diketo)
structure of the pyrimidine base, uracil, in the gas phase23
and, somewhat later, to explore the conformational landscape
of the neurotransmitter, histamine.24 Nucleic acid base structures25 as well as those of some aliphatic amino acids,26
were subsequently probed by Saykally's group at Berkeley,
through high resolution experiments conducted in the
IR using diode laser spectroscopy or the ingenious and sensitive
caity ring down technique.27,28
Pulsed, Fourier transform
microwave spectroscopy, a time domain technique initially
developed by Flygare29 and exploited by Legon,30
and
its analogue, electronic rotational coherence spectroscopy (RCS),
developed and exploited by Felker and Zewail,31 offer exciting
and so far, underemployed alternative strategies for determining
small biomolecular structures. Electron diffraction
offers another (non-spectroscopic) alternative, though analysis
of the scattering patterns becomes complicated when there
is a spread of contributing conformers.32 In the condensed
phase, the matrix isolation technique has been exploited,
both in the neutral environment of frozen rare gases33 and in the ionic environment of a crystalline salt such as KBr.34
Most recently, Toennies and Vilesov35 have used the
‘gentle’ and isothermal low temperature environment of a liquid
helium droplet to isolate individual molecular conformers
of tyrosine and tryptophan. This approach provides an enticing,
and different alternative to the collisional cooling process
in a supersonic, free-jet, rare gas expansion35–37—it is a technique
that is likely to be exploited increasingly in the near future.
Structural assignments
Until a few years ago, most gas phase structural assignments were based upon the analysis of pure rotational spectra, or the analysis and/or simulation of the rotational structure presented in fully (or near fully) Doppler-resolved molecular ro-vibronic spectra.38 They have also been based upon the rotational recurrences presented in their electronic rotational coherence spectra, recorded in the picosecond time domain,31 crucially aided in every case, by ab initio calculation. In the last few years, however, this strategy has been considerably broadened (in more ways than one!) by the initially unexpected sensitivity of partially resolved electronic rotational band contours, recorded at lower resolution, to changes in molecular conformation or supramolecular structure.39–41Fig. 3 shows a dramatic example of this in 3-phenyl propionic acid, an analogue of phenylalanine. Many other examples, which have exploited the remarkable versatility of this approach, will be encountered later on in this article.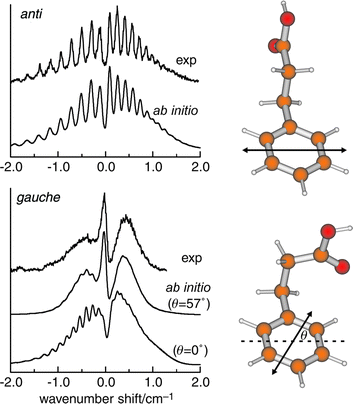 | ||
| Fig. 3 Rotational contours of the S1←S0 origin bands of 3-phenylpropionic acid at 37627 cm−1 (anti) and 37649 cm−1 (gauche), obtained by fluorescence excitation. The simulated contours are based on ab initio data for the structures shown alongside, including the calculated electronic transition moment alignments indicated by the double-headed arrow. For the lowermost trace, θ = 0° was used instead.39,40 | ||
In each of these strategies the ‘
structural images’ have been reflected
in their rotationally resolved (or partially resolved) spectra. Ultimately, of course, the rotational structures (or rotational
band contours) only reflect global properties of the molecular
conformers (or clusters). Despite the undoubted power
of rotational spectroscopy, its limitations increase with increasing
molecular complexity and/or cluster size, notwithstanding
the support provided by ab initio, or semi-empirical computation. Vibrational spectroscopy provides a complementary,
‘
tailored’, and often bond-specific strategy, which can provide
extremely valuable information about local interactions,
particularly those involving hydrogen bonding. Its realisation,
still crucially aided by ab initio computation, in the IR
(or stimulated Raman) double resonance technique, known as
mass-selected, resonant ion-dip IR (or stimulated Raman) sp
ectroscopy, RIDIRS (or SRIDS), is ideally designed for probing
the structures of small hydrogen-bonded biomolecules
and biomolecular clusters. This strategy was first developed
by Lee15 and subsequently developed and exploited by Mikami,42 Zwier,43,44 and their co-workers. The vibrational spectra
of individual, isolated molecular conformers, and/or their
size-selected clusters, are recorded ia the dips in their frequency
and mass-selected R2PI signals, or alternatively, their frequency
selected LIF spectral intensities. These occur whenever
the infrared laser radiation (or the laser ‘pump-dump’ difference
frequency, in a stimulated Raman experiment) is tuned
into resonance with a molecular vibrational transition.
The O–H, N–H or C–H stretching modes, which are particularly sensitive to H-bonded interactions, all lie in the readily accessible
mid-IR region. An illustrative example, shown in Fig.
4, displays IR bands associated with the O–H stretching modes
of the (gauche) molecular conformer of benzyl alcohol,
its (cyclic) singly hydrated cluster, and its (hydrogen-bonded)
dimer.45 The increasingly red-shifted, and broadened, O–H
bands reflect the development of hydrogen-bonded interactions
involving the alcohol side chain, the bound water molecule,
or the π-electrons on the aromatic ring.
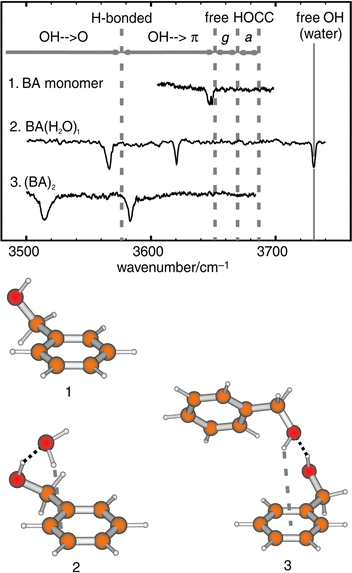 | ||
| Fig. 4 IR–UV ion depletion spectra and assigned structures for benzyl alcohol (1), its 1:1 hydrate (2) and its dimer (3). The structures shown are based on ab initio calculations at the MP2/6-31 + G**, MP2/6-31G* and HF/6-31G* levels respectively. The small red-shift for the monomer OH stretch reflects weak OH···π H-bonding. | ||
Computational
The combination of rotational structural analysis and IR (or stimulated Raman) ion-dip spectroscopy, has provided a considerably more powerful aid to molecular structural exploration than either strategy could provide alone, but the explorer would still be lost without the guidance provided by ab initio theory. Without reliable, ab initio computation quantitative analysis and interpretation of the experimental data would simply not be possible. It is the array of different, independent experimental measurements, coupled with the enormous increase in computing power, that is allowing molecular optical spectroscopists to peer with confidence, into the world o f small biomolecules and record quantitative images of their structures.The
level of theory chosen is a compromise between reliability
and computational expense, which naturally depends
on the size of the system and the number of possible structural
permutations. When many structures are possible, a typical
strategy involves generating a comprehensive set of feasible
conformers or cluster structures as a starting point and optimising
each one using the Hartree–Fock method and standard
basis sets of moderate size, e.g. 6-31G(d). The most stable structures,
i.e. those which may be expected to have significant populations
at ambient temperatures, are then re-optimised using
a larger basis set and taking electron correlation into account
(typically at the MP2 level), or alternatively, using the DFT,
Becke3LYP hybrid functional. Stationary points, characterised
by calculation of harmonic frequencies using analytical
second derivatives, yield the vibrational frequencies and provide
the zero-point energy (ZPE) corrections as well as IR and
Raman intensities. Depending upon the level of theory used
for the calculations, appropriate ‘anharmonic’ scaling factors
can be introduced to compare computed frequencies with the
experimental frequencies actually observed. More sophisticated
vibrational self-consistent field methods for achieving
this are currently being developed by Gerber.46Relatie
energies obtained in this way are fairly reliable (typically
within a few kJ mol−1), but they can be improved slightly
by single point calculations using as large a basis set as possible.
The absolute magnitude of the binding energy within a
complex is more difficult to calculate with confidence, given tha
t extremely large basis sets are required for BSSE corrections
to converge (to zero).
3. Early days
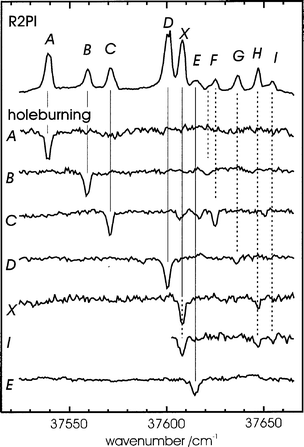
The early pioneering experiments, conducted in the UV, using laser induced fluorescence (LIF) or resonantly enhanced two-photon ionisation (R2PI) spectroscopy, particularly by Levy in Chicago,47,48 helped to lay the foundations for much of what has followed. Early targets included the aromatic amino acids, tryptophan,49 tyrosine and phenylalanine,50 and neurotransmitters such as tryptamine51 (a close relative of serotonin (5-hydroxy tryptamine) and melatonin) and 2-phenyl ethylamine.52 In 1985 and 1986, Levy's group published a flurry of papers presenting evidence that “ different conformations of tryptophan, tryptamine and other tryptophan analogues were present in the gas phase in a seeded supersonic expansion”.53 The conformers were identified through the multiplicity of band origins contributing to their S1←S0 excitation spectra, which were recorded initially, at vibrational levels of resolution, using LIF detection coupled with the saturation technique (see Fig. 5). These landmark papers were rapidly followed by others, reporting the vibrationally resolved LIF and R2PI excitation spectra of a series of tryptophan peptides, such as Trp-Gly, Gly-Trp and Trp-Gly-Gly: again they showed evidence of contributions from alternative conformers and also revealed differences between N-terminated and C-terminated isomers.54,55 A few years later, Li and Lubman reported R2PI spectra of tyrosine and its analogues using a pulsed laser desorption technique.56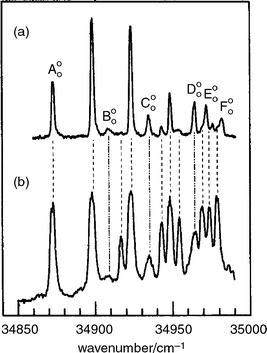 | ||
| Fig. 5 R2PI spectrum of tryptophan. The spectrum in (b) was recorded using a laser power density ca. 100 times greater than in (a). Transitions assigned to origins are labelled A00–F00. (Modified from Fig. 6 of ref. 47.) | ||
Experiments had also been conducted at much higher resolution, to record the rotationally resolved S1←S0 fluorescence excitation spectrum of the parent chromophore, indole57 and it was not long before similar spectra were obtained for each of the band origins identified in tryptamine58 and its isotopomer, tryptamine-d3.59 Their rotational analysis, guided by semi-empirical computation, led to structural assignments for most of its side-chain conformations.58,59 The assignments were subsequently extended by Felker and his co-workers,60 through analysis of the rotational recurrences in the time-resolved RCS spectra of individually selected conformers. Some of the structures, assigned to eclips ed conformations, were surprising at the time and remained so for many years until they were re-visited by Zwier.61 The latest results, obtained using IR ion-dip spectroscopy coupled with DFT computation, now establish a preference for staggered, out-of-plane conformations.
The hybrid character of the rotational structure in indole provided the angular magnitude, but not the absolute alignment of the S1←S0 transition moment (TM), referenced to the molecular inertial axes. It could be deduced however, by assuming that the TM alignment, referenced to the molecular frame, was unaffected either by the introduction of side-chain substituents or by changes in their conformation, both in tryptamine and (by implication) tryptophan. On this basis, a comparison of the TM orientations determined in indole57,62 and tryptamine,53 allowed the S1←S0 electronic transitions in both molecules to be associated with excitation into the 1Lb state. Further light was (literally) shed upon this assignment, through an analysis of the dispersed fluorescence spectra of tryptophan, generated through the selective laser excitation of each of its jet-cooled conformer bands.48 In most of them, almost all the emission was associated with resonance fluorescence but the excitation of one particular conformer also generated an intense, broad and red-shifted fluorescence, see Fig. 6. The additional emission was ascribed initially, to the formation of an excited zwitter-ion, produced through excited state proton transfer from the carboxylic acid to the neighbouring amino group along the folded amino acid side chain. Later studies, however,63 attributed the emission simply to the generation of a strong electric dipole, associated with intramolecular hydrogen bonding along the amino acid side chain which interacted with the strongly dipolar S2(1La) state of the indole chromophore to lower its energy. If the interaction were sufficient to invert the S2–S1 energy gap, it would allow electronic state mixing to promote radiationless transfer from S1 → S2; the broad, red-shifted emission was associated with fluorescent decay from S2 → S0. Significantly, the red-shifted fluorescence was not detected in the emission from the amino acid ester63 where the proposed hydrogen-bonded interaction would be blocked. Despite the array of circumstantial evidence in favour of the current analysis however, it is likely that the last word has not yet been written on the subject.64
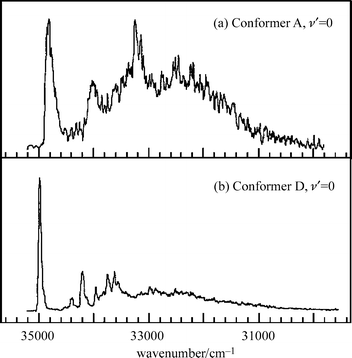 | ||
| Fig. 6 Dispersed fluorescence spectra of tryptophan subsequent to exciting the electronic origins of (a) conformer A, and (b) conformer D, as shown in Fig. 5. (Modified from Fig. 4 of ref. 48). | ||
Some years later, new studies39,40 of substituted benzene
(rather than indole) conformers, led to a reassessment of the
assumed general insensitivity of the TM orientation to
changes in the conformation of side-chain substituents.40 The first
straw in the wind was provided by a new study of 3-phenyl-1-propionic
acid,39 a near relative of the amino acid, phenylalanine,
which can adopt an extended (anti) or a folded (gauche)
conformation.39,40,65 The experimental and simulated
rotational contours of the two S1←S0 band origins, could
only be reconciled if the TM did rotate (away from the short
axis of the aromatic ring) when the side chain was folded into
its gauche conformation (see Fig. 3). The ‘normal’ TM orientation
in the extended conformer reflects the 1Lb character of
its S1
state but in the gauche conformer, a mix of ‘
through
bond’ and ‘through space’ perturbations leads to electronic
state mixing introducing a measure of 1La character into the excited
state of the aromatic chromophore.41 In substituted benzene
molecules, the S2–S1 energy gap remains too great to allow
radiationless transfer from S1
→ S2
but the incidence of TM rotation
can lead to a very useful result—a major ‘amplification
’ of the spectral consequences of conformational change, at least in substituted benzene molecules. Many other examples
were soon discovered.40,66–68 The analysis was qualified
later, by the caeat that state mixing/TM rotation can also
be promoted (or augmented) by other (interesting) processes, notably vibronic interactions,45,69 or the binding of polar
molecules to the flexible side chain.68
Until recently, despite a plethora of theoretical predictions for the binding of water at alternative sites on the amide group (the basic link in peptide and protein chains), spectroscopic measurements of hydrated amide, let alone peptide structures at the isolated molecular level, were almost non-existent: they are still rare. A similar situation prevails a fortiori for the experimental spectroscopic observation of intermolecular binding in nucleic acid base pairs.70 Pioneering studies of the cyclic amide, 2-pyridone, its hydrogen-bonded dimer and its molecular hydrates,38,71,72 particularly by Pratt and his co-workers, provided a notable exception. The molecule was very ‘user-friendly’ (in contrast to its analogue, uracil73), displaying a rotationally structured LIF excitation spectrum, a quality retained both in its singly or doubly hydrated clusters72 and in the dimer.74,75 On the other hand its use as a peptide analogue, where a trans-conformation is generally the norm (unless the amide is followed by a proline residue, when the cis- and trans-configurations have similar probabilities), was limited by its cyclic structure, which forces the amide into a cis-configuration.
That
said, in a seminal series of papers72,74–76
Pratt and his co-workers
were able to determine the cyclic structures of the two
hydrates and of the molecular dimer, using a combination of
high resolution Doppler free spectroscopy coupled with deuterium
and H218O isotopic substitution, see Fig. 7. In particular,
they were able to establish the anti-parallel hydrogen-bonded
(rather than stacked) geometry of the molecular dimer,
which generates the planar, C2h
symmetry structure74,75
both in the ground (1Ag–S0) and electronically excited
(1Bu–S2) states.‡ Hydration induces a significant distortion
of the amide group toward a zwitterionic structure, suggesting
the possibility of a keto–enol tautomeric pathway (to produce
2-hydroxy pyridine) mediated ia proton transfer along a hydrogen-bonded
water chain. Indeed the key message conveyed by this benchmark study, was the correlation between
molecular (and supramolecular) structure and the electronic
wavefunction—which could be polarised by the solute–solvent
interaction within an isolated molecular cluster. Charge separation
plays a crucial role in acid–base catalysis and in enzyme-catalysed reactions. These experiments offered the first possibility
of monitoring its onset directly, promoted through local interactions
in size-selected molecular clusters of known structure and conformation, in the absence of the additional, though no
doubt important perturbations associated with a surrounding
condensed solvent medium.
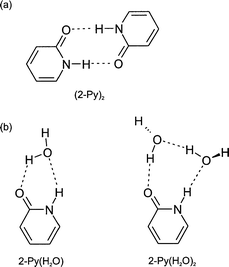 | ||
| Fig. 7 Schematic representation of the structures of (a) the 2-pyridone dimer and (b) its 1:1 and 1:2 hydrates. | ||
4. Neurotransmitters
The control of biological processes is generally mediated by small molecules which bind to specific macromolecular receptor sites. Those which promote specific responses are termed agonists; those which inhibit the response are antagonists. Molecular pharmacology is largely concerned with understanding their modes of operation and the hugely important industrial goal of controlled synthetic drug design is devoted to optimising their action (and minimising their unwanted side-effects). Molecules which mediate the connections between separate nerve cells, or between nerve and muscle cells, belong to the family of neurotransmitters. Many share a common structural motif, for example, mono-amines such as γ-amino butyric acid (GABA) or those involving aromatic, or heterocyclic rings such as phenylethylamine and dopamine (where the flexible ethylamine side chain is attached to benzene or catechol), or histamine, tryptamine and serotonin (where it is attached to an imidazole, indole or 5-hydroxy indole ring); and ethanolamines (and their derivatives) such as acetylcholine, or the ephedrine or adrenaline series where again, the side chain is attached to a benzene or catechol ring. Representative examples are shown in Fig. 8.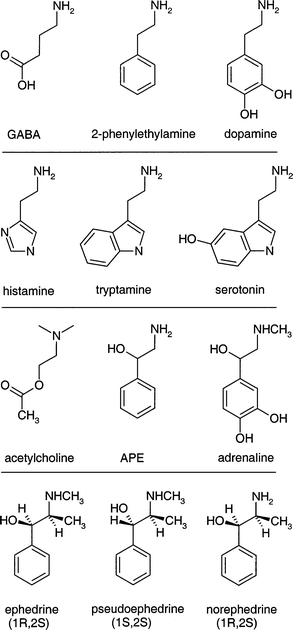 | ||
| Fig. 8 Chemical structures of a series of neurotransmitters. | ||
The notion of a common motif has led to the concept of a ‘pharmacophore ’—a common, pharmacologically active structural configuration. This concept leads in turn, to the question: how readily do the favoured molecular conformations approach the common structural configuration which may be associated with their optimum pharmacological activity and what are the determining factors in achieving this? The flexibility and conformation of their side chains (and perhaps their bio-activity) can be influenced by many potential structural controls. These may include intramolecular hydrogen bonding, covalent cross-linking (which can destroy the flexibility of the mono-amine side chain, as for example, in morphine), dispersive interactions, steric repulsions, chirality, hydrogen-bonded interactions with neighbouring molecules (especially water) or donor/acceptor groups at receptor sites, or protonation of the terminal amino groups. Not surprisingly, the combination of their ‘spectroscopic accessibility’ with the need to explore and characterise their conformational landscapes, has made them a prime target for structural investigation, both theoretically and experimentally. Although most gas phase spectroscopic experiments to date have been restricted to benchmark explorations of neutral (aromatic and heterocyclic) molecules and some of their hydrated clusters, efforts to detect and characterise their protonated structures can be anticipated in the near future.
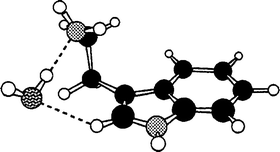
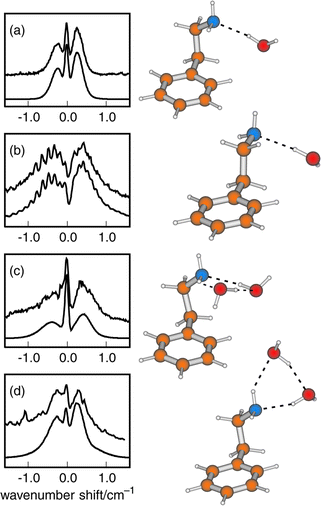
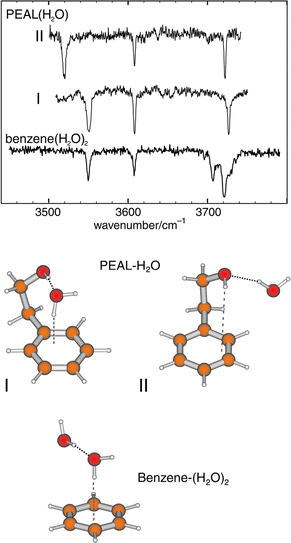
The first detailed conformational studies focused on tryptamine51 (see Section 3) and its hydrated clusters,31,77 explored through high resolution UV and rotational coherence spectroscopy, and on the two tautomers of histamine; these were characterised by Godfrey and Brown24 using a combination of microwave spectroscopy and ab initio computation. The importance of intramolecular hydrogen bonding was immediately apparent: all four of the observed conformers of histamine were assigned to folded (gauche) side-chain structures, stabilised either by hydrogen bonding from the imidazole ring to the terminal amino group, N(1)H → NH2 in the 5-substituted tautomer or more weakly, from the amino group to the ‘bare’ N(3) atom, N(3)←HNH, in the alternative, 4-substituted tautomer. Their structures are shown in Fig. 9. Hydrogen-bonded interactions are also crucial in controlling the conformational structure in the 1:1 hydrated cluster of tryptamine. Instead of the multiplicity of conformers populated in the isolated molecule, a single bound water molecule locks the amine into its most stable conformation by linking its side chain to the indole ring,31,77 see Fig. 10. The water acts as a proton donor to the amino group and is bound to the indole ring through a CringH···Owater interaction. This ‘ dual-bonding’ function is one which water is very happy to display: it will be encountered frequently.
 | ||
| Fig. 9 Ab initio structures for the four experimentally observed conformers of histamine, including: (a) 5-imidazole ethylamine and (b)–(d) 4-imidazole ethylamine. (Modified from Fig. 2 of ref. 11(b).) | ||
 | ||
| Fig. 10 Geometry of the tryptamine–water complex consistent with the rotational constants obtained from rotational coherence experiments. (Modified from Fig. 11 of ref. 31.) | ||
Intramolecular hydrogen bonding also influences the conformational preferences in 2-phenylethylamine. Its two lowest energy conformers both display folded, gauche structures—assigned initially, through microwave measurements78 and subsequently, through UV rotational band contour analysis aided by the conformationally induced rotation of the S1←S0 electronic transition moment.66 Their folded geometries allow hyd rogen-bonded interaction between the amino group and the π-electrons of the aromatic ring. Its next two conformers, which present an extended (anti) side chain and a ‘normally aligned’ transition moment, are only weakly populated.67 The Cβ-methylated derivative, amphetamine, appears to show a similar pattern of conformational behaviour.79,80 In the related molecule, 2-phenyl ethanol, the single, π-hydrogen bonded, gauche conformation is even more strongly stabilised45,66,69,81,82 and the extended, anti conformer is only a minor component in its R2PI69 and millimetre wave82 spectra.
As
in tryptamine, bound water molecules favour the preferred conformation in phenylethylamine; they also exert an
additional ‘through space’ influence on the alignment of its transition
moment.68
The supramolecular structures of its most
stable, singly and doubly hydrated conformers, determined
through rotational band contour analysis,66 are shown in
Fig. 11. In the 1:
2 clusters, the water molecules form a cyclic, hydrogen-bonded network, in which the amino binding site
can act both as a proton donor and an acceptor. This dual
behaviour is displayed even more dramatically, in the singly
hydrated complex of the analogous molecule, 2-phenyl ethanol,
where a combination of band contour analysis, IR ion-dip
spectroscopy and ab initio computation has allowed the assignment
of two equally populated, alternatie structures.69 In one, the bound water molecule inserts between the terminal
OH group and the aromatic ring to create a ‘daisy-chain’ of hydrogen bonds. These link the ethanol side chain (which acts
as a proton donor) to the water molecule, and the water to
the (proton accepting) π-electrons on the ring, to create a cyclic structure. In the alternative structure the ethanol side chain
acts as a proton acceptor, to create a non-cyclic, ‘
exo’ hydrate.
The two structures, shown in Fig. 12 generate distinct
IR spectra; the π-bonded structure and its IR spectrum, are strikingly
similar to those of the analogous benzene–(water)2 cluster,
assigned earlier by Zwier.83
 | ||
| Fig. 11 Rotational band contours of 2-phenylethylamine clusters. Upper traces: (a) fluorescence excitation spectrum of 1:1 hydrate ‘E’; (b)–(d) 2-colour R2PI (λ2 = 301 nm) spectra of 1:1 hydrate ‘ G’, and 1:2 hydrates ‘F’ and ‘H’ (see ref. 68 for band labelling). Lower traces: simulations based on ab initio data for the structures shown alongside. | ||
 | ||
| Fig. 12 IR–UV ion-depletion spectra probing the most intense 1:1 hydrate features of 2-phenylethanol.69 The spectrum of benzene–(H2O)2 adapted from ref. 83 is also shown. Assigned structures are shown below. | ||
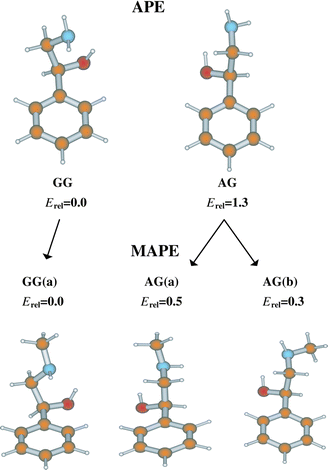
Substitution of an OH group on the Cα atom of 2-phenylethylamine,
creates the simple ethanolamine, 2-amino 1-phenyl
ethanol (APE)—the basic framework of the important class
of pharmaceuticals known as ephedra. It also introduces a chiral
centre so that each of the conformers of APE can exist
as R or S enantiomers. Their most stable structures, defined by
the arrangement (anti or gauche) of the CCCN and OCCN atomic
chains, together with those of the N-methyl derivative
(MAPE), are shown in Fig. 13. Each one is stabilised by an
intramolecular, OH → N hydrogen bond along the side-chain.
Fig. 14
displays the experimental and computed IR spectra associated
with their O–H and (anti-symmetric) N–H stretching modes;84,85
the displaced frequencies of the O–H stretch mode, red-shifted
by ca. 130 cm−1 relative to benzyl alcohol, reflect the
OH → N hydrogen-bonded interactions. Fig. 14 also shows the
IR ion-dip spectrum of the most stable singly hydrated conformer of APE. The additional bands reflect the insertion
of the water molecule between the neighbouring hydroxy and amino
groups bound by OHwater
→ N and OHAPE
→ Owaterinter-molecular
hydrogen bonds. As a result of this, the side
chain opens out to allow re-instatement of the NH →
π bonding and the
preferred molecular conformation of APE in the hydrate becomes
GG rather than AG, no longer the same as in the bare
molecule—hydration has altered its conformational landscape.
The hydrated structure can be regarded as a model biomolecular
receptor system with the amino group playing the role
of a docking site. The continuous ‘daisy-chain’ of hydrogen bonds
optimises the overall OH → water → N binding energy: it might also
help to facilitate charge transport ia proton transfer84 when this is energetically viable.
 | ||
| Fig. 13 Ab initio structures for the experimentally observed conformers of 2-amino-1-phenylethanol (APE) and N-methylamino-1-phenylethanol (MAPE). Relative energies at the MP2/6-311G** level are quoted in kJ mol−1. | ||
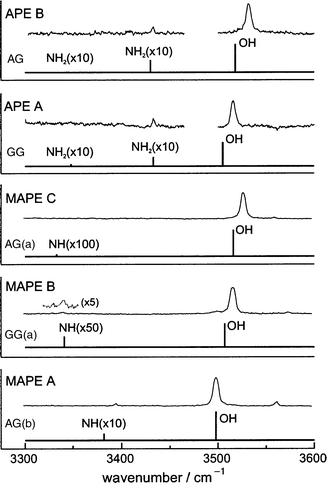 | ||
| Fig. 14 IR–UV ion-depletion spectra probing conformer origins of 2-amino-1-phenylethanol (APE) and N-methylamino-1-phenylethanol (MAPE). The accompanying stick spectra are based on B3LYP/6-31 + G* calculations on the conformers shown in Fig. 13, with OH and NH stretch frequencies scaled by 0.9734 and 0.956 respectively. | ||
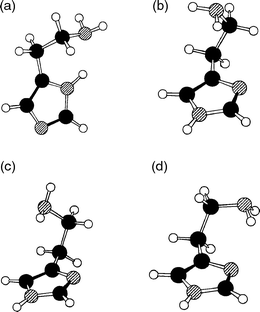
Methylation of the Cβ atom in APE, and MAPE, creates the new molecules, norephedrine and ephedrine: it also introduces a second chiral centre, to provide two pairs of physically, chemically and biochemically distinct diastereoisomeric structures, {(1R,2S), (1S,2R)} or {(1S,2S), (1R,2R)}, for each molecule. The alternative configurations create alternative local dispositions among the neighbouring groups along the side chain, which in their turn, affect the strengths of the local intramolecular interactions, both with each other and with the aromatic ring to which the side chain is attached. The end result is a spread in their individual conformational landscapes. Fig. 15 summarises the lowest energy conformations, relative energies and IR spectra of (1R,2S) ephedrine and (1S,2S) pseudo-ephedrine, computed ab initio, together with the corresponding R2PI and IR (ion dip) spectra, recorded experimentally.12 As with APE and MAPE, it is the OH → N hydrogen-bond which is the principal stabilising factor, but its length§ and strength are subtly modulated by other, weaker interactions, for example (weak) π hydrogen-bonding between the (methyl)amino group and the ring (e.g. in the two GG(a) conformers), dispersive interactions between the methyl groups and the ring (e.g. in the GG(b) conformer of pseudo-ephedrine), and steric interactions in general. The markedly different conformational landscapes in the two diastereoisomers, ephedrine and pseudo-ephedrine reflect the influence of local configuration on the fine balance of ‘ non-bonded’ interactions within the two molecules. These, in turn, influence the structure and flexibility of their side chains, at least in the gas phase.
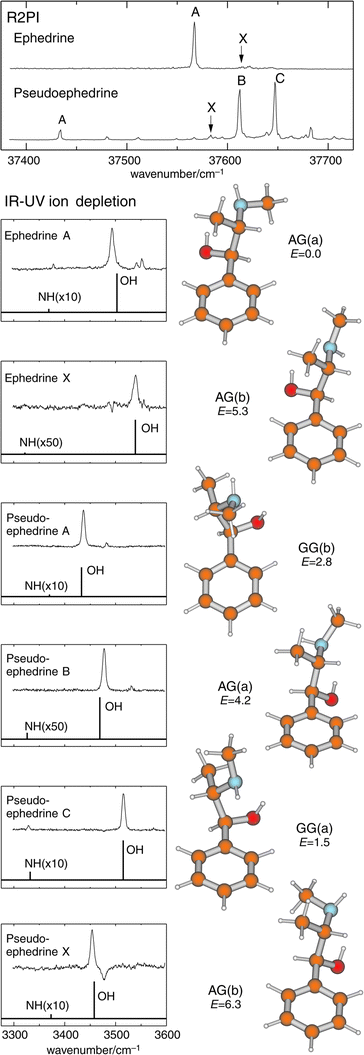 | ||
| Fig. 15 Top panel: R2PI spectra of (1R,2S) ephedrine and (1S,2S) pseudoephedrine. Lower panels: IR–UV ion-depletion spectra probing the conformer origins labelled in the R2PI spectra. Stick spectra are based on B3LYP/6-31 + G* calculations for the conformers shown alongside, with OH and NH stretch frequencies scaled by 0.9734 and 0.956 respectively. Relative energies at the MP2/6-311G** level are quoted in kJ mol−1. | ||
5. Amides and peptides
The structural, physical and biological properties of proteins are crucially dependent on their hydrogen-bonded interactions, particularly those involving the amide N–H and C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O groups.
The most important of these are the N–H···OC bonds which gove
rn secondary structure but interactions with water also warrant
special attention. Water is important not only as the bulk
solvent surrounding proteins, but as a chelating ligand found
in specific sites in their interior. IR (and Raman) spectroscopic
techniques are well suited to probing these interactions,
given the sensitivity of vibrational modes to small changes in
(hydrogen) bonding. The dependence of amide vibrations
on the local conformation, and interactions with the surrounding
solvent have particular importance given the widespread
use of IR and Raman spectroscopy as a probe of secondary
structure in peptides.86 The early success of Levy in obtaining
ultra-violet spectra of di- and tri-peptides54,55
(revisited
recently by deVries87), see Fig. 16, and the successful assignment
of the preferred conformational structure of alaninamide
by Tubergen's group88 using pulsed Fourier transform microwave
spectroscopy, also holds the promise that full conformational
and hydrated structural assignments might soon
be possible for a range of low molecular weight peptides
in the gas phase. When they accommodate aromatic residues,
a favoured approach will be a ‘
step-by-step’ strategy,
which applies to R2PI, IR–UV and UV–UV ion-dip spectroscopy,
supported by ab initio (or lower-level) computation, to
a range of small peptides of increasing complexity. While
the exponential increase in the dimensionality of the associated
conformational space might deter the faint-hearted from
such an undertaking, they may take comfort in the knowledge
that ‘non-bonded’ interactions may guide the extended peptides
into a limited number of preferred structures and eventually,
funnel them into a single global minimum.
O groups.
The most important of these are the N–H···OC bonds which gove
rn secondary structure but interactions with water also warrant
special attention. Water is important not only as the bulk
solvent surrounding proteins, but as a chelating ligand found
in specific sites in their interior. IR (and Raman) spectroscopic
techniques are well suited to probing these interactions,
given the sensitivity of vibrational modes to small changes in
(hydrogen) bonding. The dependence of amide vibrations
on the local conformation, and interactions with the surrounding
solvent have particular importance given the widespread
use of IR and Raman spectroscopy as a probe of secondary
structure in peptides.86 The early success of Levy in obtaining
ultra-violet spectra of di- and tri-peptides54,55
(revisited
recently by deVries87), see Fig. 16, and the successful assignment
of the preferred conformational structure of alaninamide
by Tubergen's group88 using pulsed Fourier transform microwave
spectroscopy, also holds the promise that full conformational
and hydrated structural assignments might soon
be possible for a range of low molecular weight peptides
in the gas phase. When they accommodate aromatic residues,
a favoured approach will be a ‘
step-by-step’ strategy,
which applies to R2PI, IR–UV and UV–UV ion-dip spectroscopy,
supported by ab initio (or lower-level) computation, to
a range of small peptides of increasing complexity. While
the exponential increase in the dimensionality of the associated
conformational space might deter the faint-hearted from
such an undertaking, they may take comfort in the knowledge
that ‘non-bonded’ interactions may guide the extended peptides
into a limited number of preferred structures and eventually,
funnel them into a single global minimum.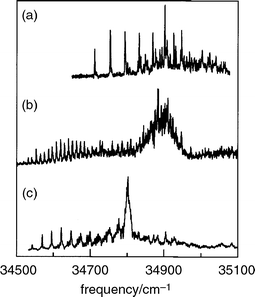 | ||
| Fig. 16 R2PI spectra of (a) Gly-Trp, (b) Trp-Gly and (c) Trp-Gly-Gly, monitoring the parent mass channel. (Modified from Fig. 2 of ref. 55.) | ||
As a first step towards the exploration of their conformational landscapes and hydrogen-bonded interactions, a series of simple amides incorporating an aromatic ring (to provide the UV chromophore) has been used to provide simple ‘ peptide models’. To date, its members have included N-phenylformamide (NPFA), and its C- and N-benzyl substituted homologues, 2-phenylacetamide (2PA) and N-benzylformamide (NBFA), together with their singly, doubly and multiply hydrated molecular clusters. Their lowest energy conformations are shown in Fig. 17.
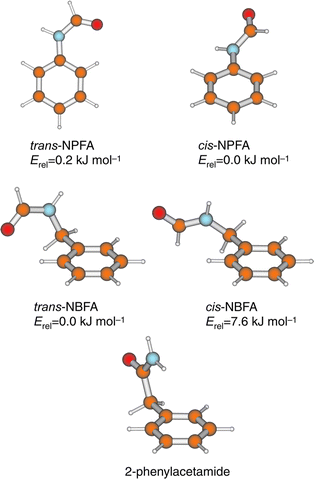 | ||
| Fig. 17 Experimentally observed conformers of N-phenylformamide (NPFA), N-benzylformamide (NBFA) and 2-phenylacetamide. Relative energies for NPFA and NBFA conformers are at the MP2/6-31G* and MP2/6-31G** levels respectively. | ||
N-phenylformamide
The cis- and trans-isomers of N-phenylformamide (NPFA) both appear in the R2PI spectrum of its seeded free jet expansion.89,90 They are readily distinguished by the vibrational and partially resolved rotational contours of their S1←S0 origin bands90 and by their distinct N–H stretch frequencies, 3441 cm−1 (cis) and 3463 cm−1 (trans), which provide another spectroscopic ‘fingerprint’ recorded through the medium of IR–UV ion-dip spectroscopy.91 The two isomers also present very different vibronic structures. cis-NPFA has a planar excited state but a ground state structure that is twisted about the phenyl–amide bond. In consequence, its S1←S0 absorption spectrum displays an extended Franck–Condon torsional progression. (A similar progression appears in the corresponding spectrum of the enzyme inhibitor, 5-phenylimidazole.92,93) In both cases their UV–UV ion-depletion ‘hole-burn’ spectra, display either the symmetric, or the antisymmetric components of the low frequency progressions, but not both. This striking behaviour provides a further diagnostic reflecting the transition from a double-minimum potential in the ground state to a single minimum potential in the coplanar, excited state.90UV
‘hole-burn’ spectroscopy also proved valuable in distinguishing
a range of singly and multiply hydrated clusters of the
trans-amide. Their structures, initially assigned through rotational
band contour analysis and subsequently confirmed through
IR ion-dip spectroscopy,90,94 are shown in Fig. 18. Clusters
with one, or two water molecules show nearly equal preferences
for binding at the N–H (proton donor) and the CO (proton
acceptor) sites. Not surprisingly, the zero-point dissociation
energies of the two 1:1 hydrates (determined from their photoionisation
and cluster-ion fragmentation thresholds) are almost identical, 23.6 ± 1.3 kJ mol−1 at the N–H site and 22.6 ± 1.2
kJ mol−1 at the CO site.95 The two di-hydrates both form cyclic
structures, with the water dimer either forming a bridge
across the terminal H–CO group or linking the amide group to the
aromatic ring ia a π-hydrogen bond. The multiply hydrated cluster incorporates
a chain of four water molecules linked across the N–H and CO sites to form a more extended hydrogen-bonded bridge.
Its ‘O–H stretch’ vibrational spectrum is characterised
by a series of broad, intense, and strongly red-shifted bands,
which reflect strong co-operative effects and a distribution of
the ‘O-H’ vibrational motion along the H-bonded chain.91 The di
scovery of this structure is particularly interesting
because hydrogen-bonded water bridges have the potential to function
as ‘proton wires’ in biomolecular assemblies. Recent theoretical
work predicts optimum proton transfer efficiencies when the bridge incorporates a chain of strong, near equivalent
and strain-free bonds96—just the kind of structure displayed by
the tetra-hydrate of trans-NPFA. Although proton transfer ia
a bridging molecular chain is not thermodynamically feasible
in NPFA it may well be implicated in proton transfers between
proteins, through trans-membrane channels, in the catalysed
formation of tautomers, or in the generation of zwitterions
in solvated amino acids (see Section 6).
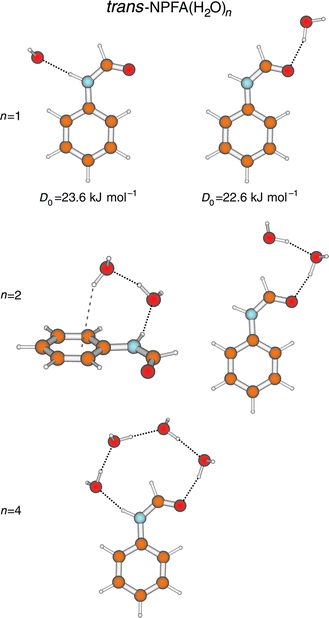 | ||
| Fig. 18 B3LYP/6-31 + G*
structures for experimentally observed hydrate clusters of trans-N-phenylformamide (NPFA). Binding energies for the 1:1 hydrates were determined ia
dissociative R2PI (see Fig. 2). | ||
N-Benzylformamide and 2-phenylacetamide
The introduction of a methylene spacer between the aromatic ring and the amide introduces a key element of flexibility into the side chain. In contrast to NPFA, the minimum energy structures of N-benzylformamide (NBFA), calculated ab initio97 and shown in Fig. 17 reveal a strong preference for the conformer which incorporates a trans-amide structure. As expected, it contributes much the more strongly in the jet-cooled S1←S0 R2PI spectrum. It can be distinguished from its nearest cis counterpart by its lower N–H stretch (3478 cm−1, cf. 3443 cm−1) and (amide I) CO overtone stretch (3435 cm−1, cf.
3465 cm−1) frequencies.97 A hetero dimer species has also
been observed, incorporating cis- and trans-NBFA units, stabilised
by NHcis···πtrans and NHtrans···OCcis
hydrogen bonds. The weak, π hydro
gen-bonded interaction, leads to a small red-shift in the N–H frequency,
28 cm−1, but the much stronger NHtrans···OC bond, which provides
an excellent model for peptide interactions, promotes
a red-shift of 102 cm−1.97 In the 2PA dimer, co-operative strengthening of the two NH···OC bonds leads to a shift of − 300 cm−1.97
This is enhanced even further in the C2h symmetry, planar dimer of 2-pyridone, where the amide group lies within the aromatic ring, and the red-shift increases to − 700 cm−1.98,99
The structures of all three dimers are shown in Fig. 7(a) and
19. Although the last example has little direct relevance for peptide–peptide interactions, it does provide a model for the
strong hydrogen-bonded interaction between uracil base pairs,74,75,98
see Section 7.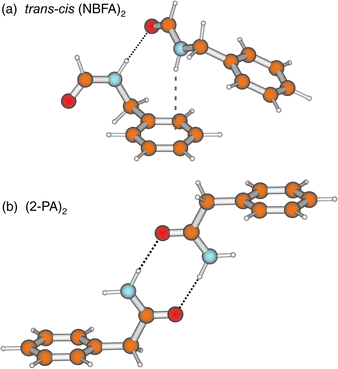 | ||
| Fig. 19 HF/6-31G* structures for the hetero dimer comprised of (a) cis- and trans-N-benzylformamide (NBFA) and (b) the dimer of 2-phenylacetamide. | ||
When the amide NBFA, is hydrated,97 the relative stability
of its cis- and trans-isomers is reversed and it is now the cis-amide
configuration, which favours the formation of strongly bonded
cyclic hydrates bridging across the amide group, that is
preferred, see Fig. 20. Its singly and doubly hydrated clusters
are bound, and stabilised, by the ‘daisy-chain’ of hydrogen-bonds
linking the neighbouring CO and N–H sites. With the
addition of a third water molecule, the cis-hydrate structure
‘flips
’. The two sites are linked through a cyclic water trimer, in preference to a linear chain and the side chain adopts a more twisted (and more energetic) conformation—a further
instance of the effect of hydration on the preferred conformation
in a flexible molecular host. In the singly hydrated trans-conformer, where the water molecule can only bind at one end of the amide group, it is bound preferentially at
the CO site, but a cyclic structure is still achieved through its secondary,
dispersive H—C···Owater interaction with the aromatic
ring. In the doubly hydrated cluster, the water dimer prefers
to bind to the N–H group instead, which allows another
bridge to be made, this time to the π-system of the aromatic ring
(cf. N-phenylformamide). Again, secondary interactions with
the ring, either dispersive, H–C···Owater, or involving π-hydrogen bonding,
O–H···πarom, play a key role in selecting the preferred
hydrate structures. As in so many other situations, it is the
secondary interactions that allow each water molecule to act both
as a hydrogen-bond acceptor and as a donor.
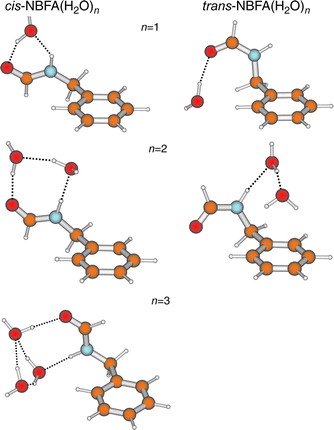 | ||
| Fig. 20 B3LYP/6-31 + G* structures for experimentally assigned hydrate clusters of cis- and trans-N-benzylformamide (NBFA). | ||
The pattern of water binding in 2-phenylacetamide100 (2PA)
is very similar to that in cis-NBFA until three water molecules
are bound when, rather than the cyclic trimer, they fo
rm a linear ‘
daisy chain’ between the N–H and CO sites. In fact, the
two structural forms are very competitive energetically, and
it is only the additional ‘weak’ CH···Owater interaction (in cis-NBFA)
that tips the balance. In general, the flexibility of the
amide side chain plays a key role in facilitating these additional
CH···Owater
interactions. When compared to the unsolvated
conformers, some of the trans-NBFA clusters exhibit considerable distortion in the dihedral angles, τ1(C2C1CαN), and τ2(C1CαNC)—equivalent
to the Ramachandran angle, φ in proteins. Peptides
consisting of glycine residues have similar flexibility to the
side chains in NBFA and 2PA.
There is a growing recognition of the importance of ‘weak interactions’ similar to those identified in the hydrated amides, in protein structures101–104 and consequently, in drug design:105,106 for example, crystallographic data reveal the Cα–H group as the most common donor in peptide CH···O contacts. CH···Owater interactions are also found when water is bound within proteins103 and carbonyl groups are common acceptors. It is possible that water molecules bound to Ci = O groups within proteins might interact with neighbouring Ci+1H groups to create an additional energy gain. The optimal value of the Ramachandran angle, φ(C1CαNC) that would allow this, φ = ± 120°, lies well within its sterically allowed range. Hopefully, it will not be too long before these types of interaction are being explored in the gas phase structures of small peptide units.64,87
6. Amino acids
Despite their familiarity and importance, the conformational landscapes of most of the natural amino acids are unknown territory. In aqueous solution or in crystalline media, they adopt charge-separated, zwitterionic structures but it is generally agreed that they all exist as ‘neutral’ molecules when isolated in the gas phase (or in rare gas matrices), as do their residues in peptide chains. In some cases this has been confirmed experimentally, e.g. through analysis of the IR gas phase spectrum of arginine;26 or valine, where IR spectra have been recorded both in the gas phase107 and in an argon matrix.108Ab initio calculations lead to the same conclusio n:109,110 the zwitterionic structure of phenylalanine, for example, is predicted to lie ca. 90 kJ mol−1 above the global minimum on the neutral potential energy hypersurface.111The
preferred conformational structures of two of the simplest
aliphatic amino acids, glycine112–114 and α-alanine,115 have been characterised through microwave and millimetre wave spectroscopy coupled with ab initio computation; they are shown
in Fig. 21. Conformer I, which corresponds to the global
minimum, benefits from a (bifurcated) hydrogen-bonded interaction
between the amino and carbonyl groups, NH2
→ OC, and
from the preferred syn-configuration of the carboxylic acid
group. In the slightly less favoured conformer, II, the syn-configuration
is sacrificed to allow the stronger hydrogen-bonded interaction
between the acidic proton and the amino group,
OH → NH2. (In principle, of course, this conformation could
lead to proton transfer and zwitterion formation, were it energetically
feasible). In the third low energy conformer, III, the
syn-configuration is recovered, permitting a weak, bifurcated hydrogen-bonded interaction between the amino and hydroxy groups,
NH2
→ OH. Although conformer III has not been detected in the gas phase, it has been identified as a trace component
in the matrix isolated IR spectra of glycine33,116,117 and
valine.108
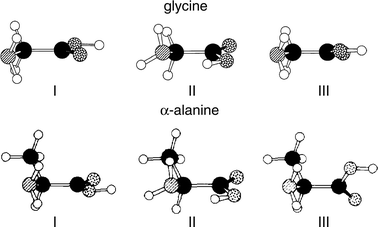 | ||
| Fig. 21 Low energy conformers of glycine (adapted from Fig. 1, in ref. 114) and alanine (adapted from Fig. 8 in ref. 11(a)). | ||
In the aromatic amino acid, β-phenylalanine, the conformational ordering, E(I)<E(II), is reversed and in addition, many more low energy conformers can be detected experimentally;50,111 UV hole-burning experiments have identified six,111 see Fig. 22. Six low-lying conformational structures are also thought to be populated in jet-cooled tryptophan49 (see Fig. 5), a conclusion confirmed by recent UV hole-burning experiments.118,119 A similar study of the R2PI spectrum of jet-cooled tyrosine has identified seven distinct band origins.120
 | ||
| Fig. 22 R2PI and UV–UV hole-burning spectra of phenylalanine, showing separate conformer origins labelled A, B, C, D, X and E. | ||
The only published, detailed structural study of an aromatic amino acid to date, has focused on β-phenylalanine.111 A combination of UV hole-burning, IR ion dip spectroscopy and high level ab initio calculation (employing large basis sets and including electron correlation), has allowed the direct assignment of five of its six lowest-lying conformers and indirectly, that of the sixth. The nine lowest energy structures calculated at the MP2 level are shown in Fig. 23; the aliphatic structures, I, II and III, identified in Fig. 20, now correspond to the aromatic side chain structures, 2 (and 8), 1 (and 3, 4) and 5. The minimum energy, hydrogen-bonded structure 1, (equivalent to II in the aliphatic series), and low energy structures 3 and 4, foster an additional stabilising interaction between the (polarised) amino group and the π-electrons on the benzene ring to generate the co-operative, ‘ daisy-chain’ sequence, OH → NH2 → π.111 The most stable conformers of tryptophan almost certainly shows the same preference63,119,121 (see Section 3) and it is likely that tyrosine will, too50,120 though this remains to be confirmed. What is already apparent, in β-phenylalanine at least, is the conformational control exerted by ‘non-bon ded’ interactions between the alanyl side chain and the aromatic ring. Seven (of the nine) most stable conformers of β-phenylalanine, adopt a folded, gauche-NCαCβC1 chain conformation, allowing π-hydrogen-bonded interactions with the amino group (cf. 2-phenyl ethyl amine). Weaker electrostatic interactions involving one of the carboxyl oxygen atoms and a neighbouring aromatic hydrogen atom also operate in conformers 1, 3, 5 and 6. Overall, conformers that are higher in energy exhibit fewer and less strong intramolecular interactions.
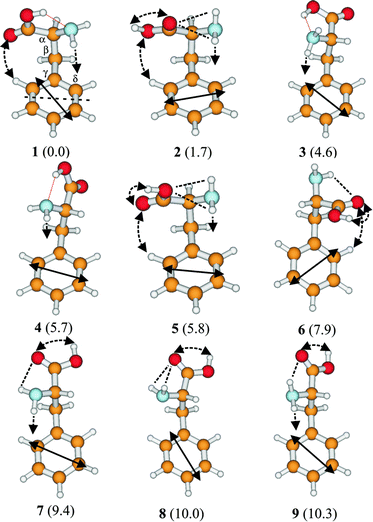 | ||
| Fig. 23 Low energy conformers of phenylalanine at the MP2/6-311G** level, with relative energies given in parentheses. Hydrogen-bonding and other interactions are shown by dotted lines and the solid, double-headed arrows indicate the electronic transition moment alignments from CIS/6-31G* calculations. | ||
The computed population distribution among the jet-cooled conformers of phenylalanine (estimated at the temperature of the pre-expansion region) corresponds quite closely to the distribution found experimentally, but only when electron correlation is taken into account. Their individual structures have been assigned primarily through analysis of the O–H and N–H stretching frequencies in their IR ion dip spectra,111 shown in Fig. 24. The O–H bands in conformers 1 and 3, which benefit from OH → NH2 hydrogen bonding, are shifted ∽300 cm−1 to the red and are strongly broadened, but in conformers 2 and 5, they remain sharp and unshifted. Subtler changes in the positions of the N–H bands, particularly the anti-symmetric stretching frequencies, are remarkably well predicted by DFT calculations. Further supporting evidence is provided by the UV R2PI rotational band contours (which are influenced by changes in the transition moment alignment, shown in Fig. 23, as well as the side-chain conformations).
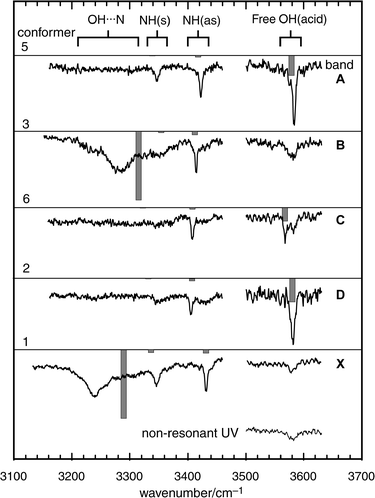 | ||
| Fig. 24 IR–UV ion-depletion spectra probing the phenylalanine conformer origins A, B, C, D and X shown in Fig. 22, together with the ion-depletion signal obtained when the UV probe laser was tuned to a non-resonant wavelength. Stick spectra are based on B3LYP/6-31 + G* calculations for conformers 5, 3, 6, 2 and 1 shown in Fig. 23, with OH and NH stretch frequencies scaled by 0.9734 and 0.956 respectively. | ||
One of the key, as yet unanswered questions concerning the amino acids, is the mechanism of their transition from a neutral to a zwitterionic structure, whether in a cluster, or in an aqueous solution, or in a crystalline environment. Much of the stabilisation in aqueous solution is provided through the solvation of the separated charges122–124 but local interactions with specific water molecules bound within the first solvation shell may also be important.125 Can a multiply hydrated, isolated amino acid cluster switch to a zwitterion? If s o, what are its preferred conformational and supramolecular structures before (and after) the switch and what is the minimum number of water molecules that is required to produce it? Ab initio calculations conducted on glycine–water clusters suggest that two are certainly not enough,122,123 and that the likely cross-over point at which the zwitterionic structure becomes the more stable, is achieved when four water molecules are bound to the amino acid126 but experimental confirmation still remains to be provided. UV bands associated with hydrated clusters of tryptophan have been detected in its S1←S0, R2PI spectra49,119,127 but their structures are, as yet, unknown. If zwitterions were produced, as they are for example, when amino acids are transferred from an aqueous to a crystalline KBr environment, their presence should be clearly signalled in their IR spectra. In a KBr matrix, the ν(O–H) and neutral ν(N–H2) stretch bands in phenylalanine and tryptophan for example, are replaced by those associated with vibration of the carboxylate and protonated amino groups.34 Within an isolated cluster the insertion of a chain of water molecules128 into the OH → NH2 hydrogen bond associated with conformer II in glycine, or conformer 1 (or 3 or 4) in phenylalanine, could provide the necessary proton wire.
7. Nucleic acid bases
Determination of the gas phase structures of natural (or substituted) nucleic acid bases and of their hydrogen-bonded interactions with each other, or with bound water molecules, or with peptide groups, has long been a prime target of research for many, very obvious reasons. The complementary, Watson–Crick pairing129,130 of the natural purine or pyrimidine bases, adenine and guanine or cytosine, uracil and thymine, determines the primary structures of DNA (T,C,A,G) and RNA (U,C,A,G). Hydration of DNA molecules plays a crucial role in promoting conformational changes and in determining their macromolecular structures in an aqueous environment.131 Protein–DNA interactions are fundamental in regulating many biological processes, including replication and expression. Direct interactions involving specific hydrogen-bonding between side chain amide amino acid groups and nucleic acid bases, have been identified in viral DNA complexes.132Unfortunately, although there is an extensive literature describing the theoretical computation of pair-wise intermolecular nucleic acid base interactions,70,133 from an experimental point of view their exploration at a molecular level has been much more an aspiration than a reality. Better news however, appears to be on the horizon with reports of new spectroscopic studies of nucleic acid base pair interactions in the IR, using the cavity ring down method28,134 and in the UV, using laser ablation and molecular beam techniques coupled with mass selected R2PI detection.135 These new developments have benefited from (a rather small number of ) earlier spectroscopic investigations of individual, non-interacting nucleic acid bases, isolated in the gas phase.
They
began with a series of Stark modulated microwave
investigations of jet-cooled uracil, thymine, cytosine and adenine
by Brown, Godfrey and their co-workers. Rotational analysis,
aided by ab initio computation, led to the assignment of
the major tautomeric (planar diketo) structures in uracil23 and
thymine,136 the two most stable (keto and enol) tautomers
of cytosine,137 and the major, N(9)H tautomer of adenine138—all are shown in Fig. 25. The diketo structure of the
predominant uracil tautomer, cooled in a free jet slit expansion,
was subsequently confirmed by Saykally25 through IR
diode laser absorption measurements of the anti-symmetric vibration
of the two CO groups. In the UV region, the gas phase S1←S0 absorption of jet-cooled uracil and its methylated derivative,
thymine, are both structureless73 suggesting rapid radia
tionless decay of the initially excited state, due perhaps, to
strong mixing of neighbouring ππ* and nπ* electronic states. In aqueous
solution both bases display very short excited state lifetimes,
ca. 10−12 s139 and very low fluorescence quantum yields,
ca. 10−4–10−6.140 Somewhat surprisingly, although the fluorescence
lifetimes and quantum yields of the purine bases,
adenine and guanine are comparable with those of their pyrimidine
counterparts (again measured in aqueous solution),
in the gas phase the S1←S0 absorption of jet-cooled guanine
presents an extensive and sharply structured vibronic spectrum141,142 (shown in Fig. 28(a)) and contributions from
the three different tautomers have been identified, with fluorescence
lifetimes ranging from 12 ns to 360 ns.141 Delayed, two-colour ionisation measurements also reveal the involvement
of a long-lived intermediate electronic state with a lifetime
>10−5 s.141 Its assignment to a triplet state accessed by intersystem crossing, while tempting, remains to be established as does
the probable (keto) tautomeric structure of the ground state.
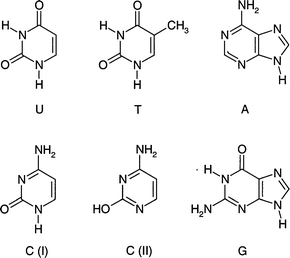 | ||
| Fig. 25 Tautomers of the DNA bases experimentally observed in the gas phase. | ||
It has been suggested that the short lifetimes of these naturally occurring, electronically excited bases in the condensed phase is no accident, but is associated with a selective evolutionary mechanism, which protects nucleic acids against photochemically induced radiation damage.143 Is it possible that the lifetimes are altered when the bases are bound through hydrogen-bonded interactions with neighbouring molecules, e.g. the amide group in a protein? An increased lifetime could increase the risk of radiation damage in the parent nucleic acid and perhaps, a mechanism for the initiation of skin cancers.
In contrast to adenine (6-amino purine), the analogue 2-amino purine, is strongly fluorescent with a quantum yield in aqueous solution, of 0.66.144 Its strong fluorescence reflects the electronic consequences of substitution at the 2-position in the six-membered ring, which shifts the energy of the ππ* state below that of the nπ* state. This makes it a very useful site-specific fluorescent probe of local oligonucleotide structure145 and a potential target for gas phase, laser ablation measurements in the future. A target which has already been triumphantly hit by this weapon, is the ribose substituted nucleoside, guanosine, and its two derivatives, 2′ and 3′-deoxyguanosine.146 R2PI spectra are shown in Fig. 26 along with the ab initio computed structures of the syn- and anti-conformers of the 3′ derivative, both of which appear to contribute to its beautifully structured R2PI spectrum. Each one is stabilised by intramolecular OH → N hydrogen bonding between the sugar and the six-membered ring.
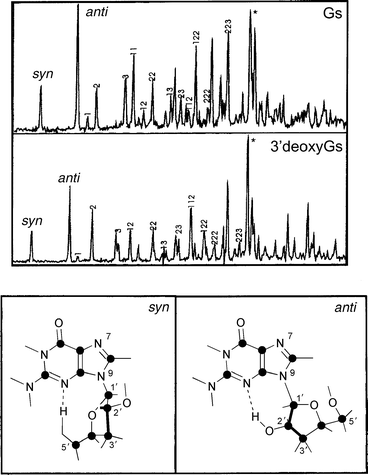 | ||
| Fig. 26 R2PI spectra of guanosine (GS) and 3′-deoxyguanosine. The syn and anti labels indicate origins of two possible conformers. HF/6-31G** structures with syn and anti conformations of 3′-deoxyguanosine are shown below (adapted from Fig. 1 and 2 of ref. 146). | ||
Experimental studies of pair-wise interactions in the gas phase
are still in the embryonic stage. Almost all of them have been
based on model systems. The planar, 2-pyridone dimer, shown
in Fig. 7(a) and discussed in Sections 3 and 5, provides an
analogue of the uracil base pair74,75,98,147 and the 2-pyridone-2-hydroxy
pyridine dimer provides a model for the reverse
keto-enol interaction.148 Femtosecond kinetic spectroscopy has revealed a sequential, tunnelling pathway for the dual H
atom exchange process in the hydrogen-bonded dimer of 7-aza-indole,149–151 providing a model for photon induced tautomer interconversion
in purine base pairs. A quantitative structure
determination still remains to be achieved152 but a schematic
representation is shown in Fig. 27(a). The structures of the singly
and doubly hydrated clusters of monomeric 7-aza-indole,
hae been assigned however, through rotational band contour
analysis of their LIF spectra152 and are shown in Fig. 27(b).
They all present cyclic hydrogen-bonded structures, bridging
across the N and NH groups. Chaban and Gordon153 have predicted, on the basis of high level ab initio
calculations,
that even the single water bridge is sufficient to produce a dram
atic decrease in the potential energy barrier to proton transfer
both in the excited and the ground electronic states. A possible
mechanism for the analogous, keto–enol transformation,
in the singly and doubly hydrated ground state complexes
of 2-pyridone72 was discussed earlier in Section 3, see Fig.
7(b). Very recently, it has been possible to move on from these
model systems, to record the first mass-selected R2PI spe
ctra of the hydrogen-bonded base pairs, guanine–cytosine and
guanine–guanine135 and a singly hydrated complex of
guanine.142 Its spectrum and predicted structure is shown in
Fig. 28 together with the structures of the most stable hydrated clusters of uracil, thymine and cytosine, computed ab initio
and also using the diffusion Monte Carlo method, by Clary
and co-workers.154
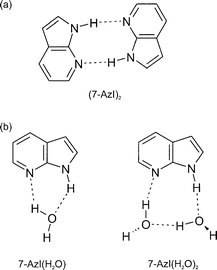 | ||
| Fig. 27 Schematic representation of the structures of (a) the 7-aza-indole dimer and (b) its 1:1 and 1:2 hydrates. | ||
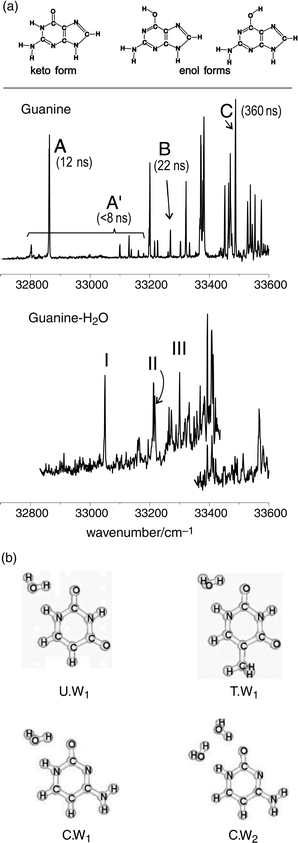 | ||
| Fig. 28 (a) UV spectroscopy of jet-cooled guanine and its 1:1 complex with water. Upper panel: Structure of the three lowest energy tautomers of guanine. Middle panel: Mass-selected R2PI spectrum including the origin transitions of three different guanine conformers (A, B and C). Evidence for the existence of these species, characterised by different excited state dynamics (fluorescence decay times in parenthesis) has been given by hole-burning spectroscopy (not shown). The same experiment shows that the weak A′ bands must be assigned to other short-lived states of conformer A. Lower panel: Mass-selected R2PI spectrum of guanine–H2O, showing three conformations (I, II, III), as revealed by hole-burning spectroscopy (not shown). (b) The most stable hydrate clusters of uracil (U.W1), thymine (T.W1) and cytosine (C.W1 and C.W2) computed by ab initio and by diffusion Monte Carlo methods (adapted from Fig. 1–4 of ref. 154). | ||
8. Future prospects
A quick trawl through the many references at the end of this review reveals a markedly bi-polar distribution. A flurry of activity in the 1980s, which first showed the exciting landscapes that were waiting to be explored; a pause for breath and ‘ consolidation’ in the early ’90s, and then in the last two or three years a flood of new research. The excitement generated by advances in chemical biology, combined with the quantitative strategies of experimental chemical physics and computational quantum chemistry is providing the classic, non-linear interaction needed for a new burst of creative research.The
‘rediscovery’ of laser ablation, coupled with free jet expansion118,120 which was introduced more than a decade ago
by Lubman,5
Levy,6 and Hunziker and de Vries7 has coincided
with the development of highly sensitive spectroscopic
strategies from the microwave to the UV, and the ready accessibility
of powerful ab initio quantum-chemical computation
codes for their interpretation. Many other techniques, which
have not been discussed in any detail here, will also make
their own contribution. The ‘pick-up’ method for preparing
single or clustered involatile biomolecules, ‘
lightly clad’ in
large helium clusters, provides a very sensitive and versatile
laboratory for their structural determination and studying
their long-range interactions, ia IR absorption spectroscopy.35
The techniques of Fourier transform and IR cavity ring
down spectroscopy provide other, so far under-exploited alternatives.
In the condensed (often aqueous) phase, resonance Raman
spectroscopy, and (in chiral systems) measurements of
the optical activity in their infra-red and Raman spectra, have
long been exploited.155 New methods of two dimensional
IR laser spectroscopy are beginning to complement NMR
spectroscopy. They offer another means of accessing biomolecular
structures, for example those of peptides, through
measurements of their amide I vibrations (mainly CO stretch)
and in particular, their couplings, revealed through
the intensities of the off-diagonal peaks156,157 (cf.
2D-NMR).
The
range of systems amenable to detailed study is also
rapidly expanding. Early reports of the mass-resolved R2PI spectra
of di- and tri-peptides,54,55,120 nucleic acid base pairs134,135
and guanosine146 have provided a foretaste of what
is to come. It should soon be possible to progress to more
complex peptides, e.g. the penta-peptide enkaphalin neurotransmitters
(which incorporate a tyrosine residue), and also
to discover how quickly the populated conformational space
in an oligo-peptide begins to ‘contract’ as the number of amino
acid residues increase. Does hydration of the amide groups
alter their trans/cis preferences and if so, the preferences for
helical s. β-sheet peptide structures? How does hydration affect
the intramolecular
hydrogen-bonded interactions between
neighbouring residues and the relative stabilities of alternative
conformations? The success in recording sharply structured
vibronic spectra in di- and tri-peptides, in using hole-burn techniques
to unravel the overlapping spectra associated with a range
of hydrated mono-amides, and in using IR ion-dip spectroscopy coupled with DFT methods to achieve their structural
assignments, encourage optimism that the answers to most,
and hopefully all of these questions will be within our sights
in the next few years.
Virtually all the biomolecules discussed in the review have been ‘neutral’, though allusion has been made to the important question of zwitterion formation, particularly in amino acids, mediated through structures which incorporate a chain of water molecules linking the proton donor and acceptor sites. Hopefully, it will not be too long before the first zwitterionic systems have been structurally assigned in the gas phase. Proton transfers are also crucially important in pathways for tautomer interconversion, for example in hydrated nucleic acid bases, with potential consequences for mutation mechanisms and in the active sites associated with catalytic triads (or diads) in serine protease enzymes. Such sites may be modelled, at least in part, by assembling termolecular, donor–imidazole–acceptor clusters in the gas phase and steps in this direction have recently been taken.92,93
The mono-amine and ethanolamine neurotransmitter structures discussed in Section 4, all reflected the neutral, unprotonated species but in a physiological environment, they are likely to be ionic. Methods already exist for recording the IR spectra of ionic clusters.17,158 New strategies to allow the preparation of protonated amines in free jet expansions and to record their IR ion-dip spectra are currently being developed. Other developments already in progress include photo-electron spectroscopic measurements of dipole-bound molecular anions, e.g. nucleic acid bases159 and indole–water clusters;160 modelling of specific drug–receptor interactions; experimental studies of chiral recognition in model enantiomeric systems especially those of pharmacological importance161–163 and measurements of intermolecular binding energies as well as supramolecular structures.95 The bridges connecting chemical physics and biology are already there—it is time to begin crossing them.
Acknowledgements
We have greatly benefited from the stimulus of collaborations and many discussions with David Pratt, Romano Kroemer and Michel Mons, tutorials from George Tranter, and crucial early and sustained encouragement from Graham Richards. We have also greatly appreciated pre-publication discussions with Richard Saykally and the receipt of pre-prints from Mattanjah de Vries and Karl Kleinermanns. We are very grateful to Lavina Snoek and Neil Macleod for their assistance in the preparation of the review and especially for the provision of some of the figures. The research that has been conducted in our laboratory has enjoyed generous support from the Leverhulme and Ramsay Trusts, the Engineering and Physical Sciences Research Council, the Laser Support Facility at the Rutherford Appleton Laboratory and GlaxoWellcome.References
- R. E. Smalley, B. L. Ramakrishna, D. H. Levy and L. Wharton, J. Chem. Phys., 1974, 61, 4363 CrossRef CAS.
- R. E. Smalley, L. Wharton and D. H. Levy, J. Chem. Phys., 1975, 63, 4977 CrossRef CAS.
- D. H. Levy, Annu. Re
![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) . Phys. Chem., 1980, 31, 197 Search PubMed.
. Phys. Chem., 1980, 31, 197 Search PubMed. - Jet Spectroscopy and Molecular Dynamics, ed. J. M. Hollas and D. Phillips, Blackie, London, 1995. Search PubMed.
- L. Li and D. M. Lubman, Re. Sci. Instrum., 1988, 59, 557 Search PubMed.
- J. R. Cable, M. J. Tubergen and D. H. Levy, J. Am. Chem. Soc., 1989, 111, 9032 CrossRef CAS; J. W. Elam and D. H. Levy, J. Phys. Chem. B, 1998, 102, 8113 CrossRef CAS.
- G. Meijer, M. S. de Vries, H. E. Hunzicker and H. R. Wendt, Appl. Phys. B, 1990, 51, 395.
- R. T. T. Karaiste, I. M. Atkinson, J. A. Shorter, A. E. W. Knight and F. R. Keene, Anal. Chem., 1993, 65, 2776 CrossRef CAS.
- R. S. Ruoff, T. D. Klots, T. Emilsson and H. S. Gutowsky, J. Chem. Phys., 1990, 93, 3142 CrossRef CAS.
- P. Felder and H. H. Günthard, Chem. Phys., 1982, 71, 9 CrossRef CAS.
- (a) P. D. Godfrey, R. D. Brown and F. M. Rogers, J. Mol. Struct., 1996, 376, 65 CrossRef CAS; (b) P. D. Godfrey and R. D. Brown, J. Am. Chem. Soc., 1998, 120, 10724 CrossRef CAS.
- P. Butz, R. T. Kroemer, N. A. Macleod and J. P. Simons, J. Phys. Chem. A, 2000, 104, in press.
- T. R. Rizzo, Y. D. Park, L. A. Peteanu and D. H. Levy, J. Chem. Phys., 1985, 85, 4819 CrossRef.
- R. J. Lipert and S. D. Colson, Chem. Phys. Lett., 1989, 161, 303 CrossRef CAS.
- R. H. Page, Y. R. Shen and Y. T. Lee, J. Chem. Phys., 1988, 88, 4621 CrossRef CAS; R. H. Page, Y. R. Shen and Y. T. Lee, J. Che m. Phys., 1988, 88, 5362 CrossRef CAS.
- T. S. Zwier, Annu. Re. Phys. Chem., 1996, 47, 205 Search PubMed.
- T. Ebata, A. Fujii and N. Mikami, Int. Re. Phys. Chem., 1998, 17, 321 Search PubMed.
- A. Courty, M. Mons, I. Dimicoli, F. Piuzzi, V. Brenner and P. Millié, J. Phys. Chem. A, 1998, 102, 4890 CrossRef CAS.
- J. E. Kenny, D. V. Brumbaugh and D. H. Levy, J. Chem. Phys., 1979, 71, 4757 CrossRef CAS.
- T. Bürgi, T. Droz and S. Leutwyler, Chem. Phys. Lett., 1994, 225, 3511; T. Bürgi, T. Droz and S. Leutwyler, Chem. Phys. Lett., 1995, 246, 291 CrossRef.
- R. D. Suenram and F. J. Lovas, J. Mol. Spectrosc., 1978, 72, 372 CrossRef CAS.
- R. D. Brown, P. D. Godfrey, J. W. V. Storey and M. P. Bassez, J. Chem. Soc. Chem. Commun., 1978, 547 RSC.
- R. D. Brown, P. D. Godfrey, D. McNaughton and A. Pierlot, J. Am. Chem. Soc., 1988, 110, 2329 CrossRef CAS.
- B. Vogelsanger, P. D. Godfrey and R. D. Brown, J. Am. Chem. Soc., 1991, 113, 7864 CrossRef CAS.
- M. R. Viant, R. S. Fellers, R. P. McLaughlin and R. J. Saykally, J. Chem. Phys., 1995, 103, 9502 CrossRef CAS.
- L. J. Chapo, J. B. Paul, R. A. Provencal, K. Roth and R. J. Saykally, J. Am. Chem. Soc., 1998, 120, 12956 CrossRef CAS.
- J.
J. Scherer, J. B. Paul, A. O'Keefe and R. J. Saykally, Chem. Re., 1997, 97, 25 Search PubMed.
- R. J. Saykally and D. Wagner, Chem. Br., July, 2000. Search PubMed.
- T. J. Balle, E. J. Campbell, M. R. Keene and W. H. Flygare, J. Chem. Phys., 1979, 71, 2723 CrossRef CAS.
- A. C. Legon, Chem. Soc. Re., 1993, 22, 153 Search PubMed.
- P. M. Felker, J. Phys. Chem., 1992, 96, 7844 CrossRef CAS.
- L. Schafer, I. S. Bin Drees, R. F. Frey, C. Van Alsenoy and J. D. Ewbank, J. Mol. Struct. (Theochem), 1995, 338, 71 CrossRef.
- I. D. Reva, A. M. Plokhotnichenko, S. G. Stepanian, A. Y. Ivanov, E. D. Radchenko, G. G. Sheina and Y. D. Blagoi, Chem. Phys. Lett., 1995, 232, 141 CrossRef CAS; I. D. Reva, A. M. Plokhotnichenko, S. G. Stepanian, A. Y. Ivanov, E. D. Radchenko, G. G. Sheina and Y. D. Blagoi, Chem. Phys. Lett., 1995, 235, 617 CrossRef CAS.
- X. Cao and G. Fischer, J. Phys. Chem. A, 1999, 103, 9995 CrossRef CAS.
- A. Lindinger, J. P. Toennies and A. F. Vilesov, J. Chem. Phys., 1999, 110, 1429 CrossRef CAS.
- K. K. Lehmann and G. Scoles, Science, 1998, 279, 2065 CrossRef CAS.
- K. Nauta and R. E. Miller, Science, 2000, 287, 293 CrossRef CAS.
- D. W. Pratt, Annu. Re. Phys. Chem., 1998, 49, 481 Search PubMed.
- P. W. Joireman, R. T. Kroemer, D. W. Pratt and J. P. Simons, J. Chem. Phys., 1996, 105, 6075 CrossRef CAS.
- J. A. Dickinson, P. W. Joireman, R. W. Randall, E. G. Robertson and J. P. Simons, J. Phys. Chem. A, 1997, 101, 513 CrossRef CAS.
- R. T. Kroemer, K. R. Liedl, J. A. Dickinson, E. G. Robertson, J. P. Simons, D. R. Borst and D. W. Pratt, J. Am. Chem. Soc., 1998, 120, 12573 CrossRef CAS.
- S. Tanabe, T. Ebata, M. Fujii and N. Mikami, Chem. Phys. Lett., 1993, 215, 347 CrossRef CAS.
- R. N. Pribble and T. S. Zwier, Science, 1994, 265, 75 CrossRef CAS.
- R. N. Pribble and T. S. Zwier, Faraday Discuss. Chem. Soc., 1994, 97, 229 RSC.
- M. Mons, E. G. Robertson and J. P. Simons, J. Phys. Chem. A, 2000, 104, 1430 CrossRef CAS.
- G. M. Chaban, J.-O. Jung and R. B. Gerber, J. Chem. Phys., 1999, 111, 1823 CrossRef CAS; G. M. Chaban, J.-O. Jung and R. B. Gerber, J. Phys. Chem. A, 2000, 104, 2772 CrossRef CAS.
- T. R. Rizzo, Y. D. Park, L. A. Peteanu and D. H. Levy, J. Chem. Phys., 1986, 84, 2534 CrossRef CAS.
- T. R. Rizzo, Y. D. Park and D. H. Levy, J. Chem. Phys., 1986, 85, 6945 CrossRef CAS.
- L. A. Peteanu and D. H. Levy, J. Phys. Chem., 1988, 92, 6554 CrossRef CAS.
- S. J. Martinez III, J. C. Califano and D. H. Levy, J. Mol. Spectrosc., 1992, 156, 225.
- Y. D. Park, T. R. Rizzo, L. A. Peteanu and D. H. Levy, J. Chem. Phys., 1986, 84, 6539 CrossRef CAS.
- S. J. Martinez, J. C. Alfano and D. H. Levy, J. Mol. Spectrosc., 1993, 158, 82 CrossRef CAS.
- L. A. Philips and D. H. Levy, J. Phys. Chem., 1986, 90, 4921 CrossRef CAS.
- J. R. Cable, M. J. Tubergen and D. H. Levy, J. Am. Chem. Soc., 1987, 109, 6198 CrossRef CAS; J. R. Cable, M. J. Tubergen and D. H. Levy, J. Am. Chem. Soc., 1988, 110, 7349 CrossRef CAS.
- J. R. Cable, M. J. Tubergen and D. H. Levy, Faraday Discuss. Chem. Soc., 1988, 86, 143 RSC.
- L. Li and D. M. Lubman, Appl. Spectrosc., 1988, 42, 418 Search PubMed.
- L. A. Philips and D. H. Levy, J. Chem. Phys., 1986, 85, 1327 CrossRef CAS.
- L. A. Philips and D. H. Levy, J. Chem. Phys., 1988, 89, 85 CrossRef CAS.
- Y. R. Wu and D. H. Levy, J. Chem. Phys., 1989, 91, 5278 CrossRef CAS.
- L. L. Connell, T. C. Corcoran, P. W. Joireman and P. M. Felker, Chem. Phys. Lett., 1990, 166, 510 CrossRef CAS.
- J. R. Carney and T. S. Zwier, J. Phys. Chem. A, 2000, 104, 8677 CrossRef CAS.
- G. Berden, W. L. Meerts and E. Jalviste, J. Chem. Phys., 1995, 103, 9596 CrossRef CAS.
- M. J. Tubergen, J. R. Cable and D. H. Levy, J. Chem. Phys., 1990, 92, 51 CrossRef CAS.
- K. W. Short and P. R. Callis, J. Chem. Phys., 2000, 113, 5235 CrossRef CAS.
- S. J. Martinez III, J. C. Alfano and D. H. Levy, J. Mol. Spectrosc., 1991, 145, 100 CrossRef.
- M. R. Hockridge, S. M. Knight, E. G. Robertson, J. P. Simons, J. McCombie and M. Walker, Phys. Chem. Chem. Phys., 1999, 1, 407 RSC.
- J. A. Dickinson, M. R. Hockridge, R. T. Kroemer, E. G. Robertson, J. P. Simons, J. McCombie and M. Walker, J. Am. Chem. Soc., 1998, 120, 2622 CrossRef CAS.
- M. R. Hockridge and E. G. Robertson, J. Phys. Chem. A, 1999, 103, 3618 CrossRef CAS.
- M. Mons, E. G. Robertson, L. C. Snoek and J. P. Simons, Chem. Phys. Lett., 1999, 310, 423 CrossRef CAS.
- K. Muller-Dethlefs and P. Hobza, Chem. Re., 2000, 100, 143 Search PubMed.
- M. R. Nimlos, D. F. Kelley and E. R. Bernstein, J. Phys. Chem., 1989, 93, 643 CrossRef CAS.
- A. Held and D. W. Pratt, J. Am. Chem. Soc., 1993, 115, 9708 CrossRef CAS.
- B. B. Brady, L. A. Peteanu and D. H. Levy, Chem. Phys. Lett., 1988, 147, 538 CrossRef CAS.
- A. Held and D. W. Pratt, J. Am. Chem. Soc., 1990, 112, 8629 CrossRef CAS.
- A. Held and D. W. Pratt, J. Chem. Phys., 1992, 96, 4869 CrossRef CAS.
- A. Held, B. B. Champagne and D. W. Pratt, J. Chem. Phys., 1989, 95, 8732 CrossRef CAS.
- L. A. Peteanu and D. H. Levy, J. Phys. Chem., 1988, 9 2, 6554 CrossRef CAS.
- P. D. Godfrey, L. D. Hatherley and R. D. Brown, J. Am. Chem. Soc., 1995, 117, 8204 CrossRef CAS.
- J. Yao, H. S. Im, M. Foltin and E. R. Bernstein, J. Phys. Chem. A, 2000, 104, 6197 CrossRef CAS.
- J. J. Urban, C. W. Cronin, R. R. Roberts and G. R. Famini, J. Am. Chem. Soc., 1997, 119, 12292 CrossRef CAS.
- R. N. Jorissen and R. D. Brown, J. Phys. Chem. A, 1999, 103, 7621 CrossRef.
- R. D. Brown and P. D. Godfrey, J. Phys. Chem. A, 2000, 104, 5742 CrossRef CAS.
- S. Y. Fredericks, K. D. Jordan and T. S. Zwier, J. Phys. Chem., 1996, 100, 7810 CrossRef CAS.
- R. J. Graham, R. T. Kroemer, M. Mons, E. G. Robertson, L. C. Snoek and J. P. Simons, J. Phys. Chem. A, 1999, 103, 9706 CrossRef CAS.
- P. Butz, R. T. Kroemer, N. A. Macleod, E. G. Robertson and J. P. Simons, J. Phys. Chem. A, 2000, 104, in press.
- J. C. Austin, T. Jordan and T. G. Spiro, in Biomolecular Spectroscopy, ed. R. J. H. Clark and R. E. Hester, Wiley, Chichester, 1993, Part A, p. 55. Search PubMed.
- R. Cohen, B. Brauer, E. Nir, L. Grace and M. S. de Vries, J. Phys. Chem. A, 2000, 104, 6351 CrossRef CAS.
- R. J. Lavrich, J. O. Farrar and M. J. Tubergen, J. Phys. Chem. A, 1999, 103, 4659 CrossRef CAS.
- V. P. Manea, K. J. Wilson and J. R. Cable, J. Am. Chem. Soc., 1997, 119, 2033 CrossRef CAS.
- J. A. Dickinson, M. R. Hockridge, E. G. Robertson and J. P. Simons, J. Phys. Chem. A, 1999, 103, 6938 CrossRef CAS.
- E. G. Robertson, Chem. Phys. Lett., 2000, 325, 299 CrossRef CAS.
- M. R. Hockridge, E. G. Robertson and J. P. Simons, Chem. Phys. Lett., 1999, 302, 538 CrossRef CAS.
- M. R. Hockridge, E. G. Robertson, J. P. Simons, D. R. Borst, T. M. Korter and D. W. Pratt, Chem. Phys. Lett., 2001, in press CrossRef CAS.
- A. V. Fedorov and J. R. Cable, J. Phys. Chem. A, 2000, 104, 4943 CrossRef CAS.
- M. Mons, I. Dimicoli, B. Tardivel, F. Piuzzi, E. G. Robertson and J. P. Simons, Chem. Phys. Lett., 2001, in press CrossRef CAS.
- Z. Smedarchina, W. Siebrand, A. Ferdandez-Ramos, L. Gorb and J. Leszczynski, J. Chem. Phys., 2000, 112, 566 CrossRef CAS.
- E. G. Robertson, M. R. Hockridge, P. D. Jelfs and J. P. Simons, J. Phys. Chem. A, 2001, in press CrossRef CAS.
- Y. Matsuda, T. Ebata and N. Mikami, J. Chem. Phys., 1999, 110, 8397 CrossRef CAS.
- Y. Matsuda, T. Ebata and N. Mikami, J. Chem. Phys., 2000, 113, 573 CrossRef CAS.
- E. G. Robertson, M. R. Hockridge, P. D. Jelfs and J. P. Simons, Phys. Chem. Chem. Phys., submitted. Search PubMed.
- G. F. Fabiola, S. Krishnaswamy, V. Nagarajan and V. Pattabhi, Acta Crystallogr. Sect. D, 1997, 53, 316 CrossRef.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991, ch. 10. Search PubMed.
- W. Saenger and T. Steiner, J. Am. Chem. Soc., 1993, 115, 4540 CrossRef.
- R. Vargas, J. Garza, D. A. Dixon and B. P. Hay, J. Am. Chem. Soc., 2000, 122, 4750 CrossRef CAS.
- D. Bradley, New Scientist, 1996, 22, 2059 Search PubMed.
- R. A. Engh, H. Brandstetter, G. Sucher, A. Eichinger, U. Baumann, W. Bode, R. Huber, T. Poll, R. Rudolph and W. von der Saal, Structure, 1996, 4, 1353 CAS.
- R. J. Saykally, personal communication..
- S. G. Stepanian, I. D. Reva, E. D. Radchenko and L. Adamowicz, J. Phys. Chem. A, 1999, 103, 4404 CrossRef CAS.
- Y. Ding and K. Krogh-Jesperson, Chem. Phys. Lett., 1992, 199, 261 CrossRef CAS.
- J. H. Jensen and M. S. Gordon, J. Am. Chem. Soc., 1995, 117, 8159 CrossRef CAS.
- L. C. Snoek, E. G. Robertson, R. T. Kroemer and J. P. Simons, Chem. Phys. Lett., 2000, 321, 49 CrossRef CAS.
- F. J. Lovas, Y. Kawashima, J.-U. Grabow, R. D. Suenram, G. T. Fraser and E. Hirota, J. Astrophys., 1995, 455, L201 Search PubMed.
- P. D. Godfrey and R. D. Brown, J. Am. Chem. Soc., 1995, 117, 2019 CrossRef CAS.
- S. J. McGlone, P. S. Elmes, R. D. Brown and P. J. Godfrey, J. Mol. Struct., 1999, 486, 225 CrossRef.
- P. D. Godfrey, S. Firth, L. D. Hatherley, R. D. Brown and A. P. Pierlot, J. Am. Chem. Soc., 1993, 115, 9687 CrossRef CAS.
- S. G. Stepanian, I. P. Reva, E. D. Radchenko, M. T. S. Rosado, M. L. T. S. Duarte, R. Fausto and L. Adamowicsz, J. Phys. Chem. A, 1998, 102, 1041 CrossRef CAS.
- F. Huisken, O. Werkahn, A. Y. Ivanov and S. A. Krasnokutski, J. Chem. Phys., 1999, 111, 2978 CrossRef CAS.
- F. Piuzzi, L. Dimicoli, M. Mons, B. Tardivel and Q. Zhao, Chem. Phys. Lett., 2000, 320, 282 CrossRef CAS.
- L. C. Snoek, M. R. Hockridge, R. T. Kroemer and J. P. Simons, in preparation..
- R. Cohen, B. Brauer, E. Nir, L. Grace and M. S. de Vries, J. Phys. Chem. A, 2000, 104, 6351 CrossRef CAS.
- R. A. Engh, L. X.-Q. Chen and G. R. Fleming, Chem. Phys. Lett., 1986, 126, 365 CrossRef CAS.
- J. H. Jensen and M. S. Gordon, J. Am. Chem. Soc., 1995, 117, 8159 CrossRef CAS.
- J. H. Jensen and M. S. Gordon, Acc. Chem. Res., 1996, 29, 536 CrossRef.
- M. Nagaoka, N. Okuyama-Yoshida and T. Yamabe, J. Phys. Chem. A, 1998, 102, 8202 CrossRef CAS.
- E. Tajkhorshid, K. J. Jalkanen and S. Suhai, J. Phys. Chem. B, 1998, 102, 5899 CrossRef CAS.
- R. T. Kroemer, ab initio density functional theoretical calculations of glycine–water clusters, personal communication..
- C. K. Teh, J. Sipior and M. Sulkes, J. Phys. Chem., 1989, 93, 5393 CrossRef CAS.
- A. Fernandez-Ramos, Z. Smedarchina, W. Siebrand, M. Z. Zgierski and M. A. Rios, J. Am. Chem. Soc., 1999, 121, 6280 CrossRef CAS.
- J. D. Watson and F. H. Crick, Nature (London), 1953, 171, 964 CAS.
- M. D. Topal and J. R. Fresco, Nature (London), 1976, 263, 285 CAS.
- W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984. Search PubMed.
- R. S. Hedge, S. R. Grossman, L. A. Lainins and P . B. Sigler, Nature, 1992, 359, 505 CrossRef.
- P. Hobza and J. Sponer, Chem. Re., 1999, 99, 3247 Search PubMed.
- R. J. Saykally and D. Wagner, Chem. Br., personal communication. Search PubMed.
- E. Nir, K. Kleinermanns and M. S. de Vries, personal communication..
- R. D. Brown, P. D. Godfrey, D. McNaughton and A. Pierlot, J. Chem. Soc., Chem. Commun., 1989, 37 RSC.
- R. D. Brown, P. D. Godfrey, D. McNaughton and A. Pierlot, J. Am. Chem. Soc., 1989, 111, 2308 CrossRef CAS.
- R. D. Brown, P. D. Godfrey, D. McNaughton and A. Pierlot, Chem. Phys. Lett., 1989, 156, 61 CrossRef CAS.
- A. Reuther, H. Iglev, R. Laenen and A. Lambereau, Chem. Phys. Lett., 2000, 325, 360 CrossRef CAS.
- P.
R. Callis, Annu. Re. Phys. Chem., 1983, 34, 329 Search PubMed.
- E. Nir, L. Grace, B. Brauer and M. S. de Vries, J. Am. Chem. Soc., 1999, 121, 4896 CrossRef CAS.
- F. Piuzzi, Q. Zhao, M. Mons, B. Tardivel and I. Dimicoli, in preparation..
- J. Cadet and P. Vigny, in Bioorganic Photochemistry, ed. H. Morrison, Wiley, New York, 1990, vol. 1, p. 1. Search PubMed.
- A. Holmén, B. Nordén and B. Albinsson, J. Am. Chem. Soc., 1997, 119, 3114 CrossRef CAS.
- J. M. Jean and K. B. Hall, J. Phys. Chem. A, 2000, 104, 1930 CrossRef CAS.
- E. Nir, P. Imhof, K. Kleinermanns and M. S. de Vries, J. Am. Chem. Soc., 2000, 122, 8091 CrossRef CAS.
- A. Müller, F. Talbot and S. Leutwyler, J. Chem. Phys., 2000, 112, 3717 CrossRef CAS.
- S. Leutwyler, personal communication..
- A. Douhal, S. K. Kim and A. H. Zewail, Nature, 1995, 278, 260 CrossRef CAS.
- A. Douhal, M. Moreno and J. M. Lluch, Chem. Phys. Lett., 2000, 324, 75, 81.
- D. E. Folmer, E. S. Wisniewski and A. W. Castleman, Chem. Phys. Lett., 2000, 318, 637 CrossRef CAS.
- A. Nakajima, M. Hirano, R. Hasumi, K. Kaya, H. Watanabe, C. C. Carter, J. M. Williamson and T. A. Miller, J. Phys. Chem. A, 1997, 101, 392 CrossRef CAS.
- G. M. Chaban and M. S. Gordon, J. Phys. Chem. A, 1999, 103, 185 CrossRef CAS.
- T. van Mourik, D. M. Benoit, S. L. Price and D. C. Clary, Phys. Chem. Chem. Phys., 2000, 2, 1281 RSC.
- Faraday Discuss. Chem. Soc., 1994, 99. Search PubMed.
- P. Hamm, M. Lim, W. F. de Grado and R. M. Hochstrasser, Proc. Natl. Acad. Sci. USA, 1999, 96, 2036 CrossRef CAS.
- S. Woutersen and P. Hamm, J. Phys. Chem. B, 2000, 104, 11316 CrossRef CAS.
- L. I. Yeh, Y. T. Lee and J. T. Hougen, J. Mol. Spectrosc., 1994, 164, 473 CrossRef CAS.
- C. Desfrancois, W. Abdoul-Carime, S. Carles, V. Périquet, J. P. Schermann, D. M. A. Smith and L. Adamaowicsz, J. Chem. Phys., 1999, 110, 11876 CrossRef CAS.
- S. Carles, C. Desfrancois, J. P. Schermann, D. M. A. Smith and L. Adamowicsz, J. Chem. Phys., 2000, 112, 3726 CrossRef CAS.
- A. Al Rasbaa, K. Le Barbu, F. Lahmani and A. Zehnacker-Rentien, J. Phys. Chem. A, 1997, 101, 3273 CrossRef.
- K. Le Barbu, V. Brenner, Ph. Millié, F. Lahmani and A. Zehnacker-Rentien, J. Phys. Chem. A, 1997, 102, 128 CrossRef CAS.
- F. Lahmani, K. Le Barbu and A. Zehnacker-Rentien, J. Phys. Chem. A, 1999, 103, 1991 CrossRef CAS.
Footnotes |
| † Present address: Chemistry Department, Monash University, Clayton 3168, Australia. |
| ‡ Access to the S state is forbidden by the g↔u selection rule. |
| § Typically lying in the range 210–220 pm but with highly non-linear ∠OH–N bond angles ∽120°. |
| This journal is © the Owner Societies 2001 |