Photoprocesses of thiamonomethinecyanine monomers and dimers
Alexander K.
Chibisov
a,
Galina V.
Zakharova
a and
Helmut
Görner
b
aCenter of Photochemistry, Russian Academy of Sciences, 117421, Moscow, Russia
bMax-Planck-Institut für Strahlenchemie, D-45413, Mülheim an der Ruhr, Germany
First published on 7th December 2000
Abstract
The photophysical properties and the monomer–dimer equilibrium of four thiamonomethinecyanine dyes (1–4) were studied by time-resolved and steady-state spectroscopic methods. In ethanol and aqueous solution at room temperature the behaviour is mainly due to a monomer and dimer, respectively. The dimerization constant for 2, the triethylammonium salt of 3,3′-di-(γ-sulfopropyl)-4,5-benzo-5′-chlorothiamonomethinecyaninebetaine, in aqueous solution is KD = 7 × 105 M−1 and lower in the presence of ethanol. Fluorescence occurs from both monomer and dimer; the quantum yield is Φf⩽0.006 for 1–4 in ethanol and 10–20 times higher in aqueous solution. The triplet state of the dimers was characterized; the lifetime is 0.1–0.9 ms and the yield ⩽0.2. With increasing temperature the amount of dimer is reduced, resulting in a correspondingly smaller value for T–T absorption. In microheterogeneous media, e.g. cetyltrimethylammonium bromide (CTAB), sodium dodecyl sulfate (SDS) or Triton X-100, the equilibrium is shifted at 25°C towards the solubilized monomer above the critical micellization concentration (c.m.c). The CTAB concentrations for dimer deaggregation of 1, 3 and 4 are much lower (10–30 μM) than the c.m.c. The amount of T–T absorption decreases with the CTAB concentration and resembles that of the dimer ground state. For 2 the dimers are converted into J-aggregates at [CTAB] in the 10–20 μM range and split into monomers around the c.m.c.
1 Introduction
The study of the photophysical processes and photochemical reactions of aggregated molecules attracts much attention and is the subject of many publications.1–24 Aggregated molecules of cyanine dyes play a key role in spectral sensitization.1 With increasing cyanine concentration monomers form dimers before formation of higher aggregates occurs.2,6 Shifting the monomer–dimer equilibrium of dyes in aqueous solution may cause changes in the overall quantum yields of fluorescence (Φf) and intersystem crossing (Φisc). Monomers of rhodamine dyes have a high Φf and low Φisc, while their aggregates reveal a strong enhancement of Φisc.3,4 Triplet state formation has been reported for dimers of various compounds7–14 and pigments in reaction centers of photosynthesis.15 Dimerization of cyanine dyes can be enhanced in the presence of salts16 as well as by the association with hydrophobic borate anions17 and can change the lifetime of the excited singlet state.18,19 J-aggregates of cyanine dyes show non-linear optical properties.20 For 5,5′-dichlorothiamonomethinecyaninebetaine and 4,5-benzothiamonomethinecyaninebetaine (1) in aqueous solution the thermodynamic parameters of dimerization have been reported.21,22For thiacarbocyanine dyes in aqueous solution non-fluorescent dimers with a face-to-face alignment and a relatively high Φisc have been suggested in contrast to monomeric dyes exhibiting low Φisc.23 Moreover, using sodium dodecyl sulfate (SDS), cetyltrimethylammonium bromide (CTAB) and Triton X-100 as anionic, cationic and non-ionic surfactants, respectively, we have recently reported on the effects in aggregation–deaggregation processes.24 With increasing surfactant concentration the monomer and dimer absorption reached 1/2 of their maximum values (conc1/2) at the critical micellization concentration (c.m.c) for similarly charged reactants. This is in contrast to oppositely charged reactants, where H- and J-aggregates appear at conc1/2 values much smaller than the c.m.c. The fluorescence intensity reflects the behaviour of the monomer, whereas the amount of T–T absorption reflects that of the dimer.24
In this paper we present results concerning a monomer–dimer equilibrium of four thiamonomethinecyanine dyes (1–4)
in aqueous and ethanol solutions. These dyes are generally
used for spectral sensitization of AgCl emulsions at 400–500
nm, where the intrinsic sensitivity of AgCl grains is very low.6
This work attempts to investigate the role of excited dimers of
water-soluble dyes in the deactivation processes. The triplet
state of the dimer was examined by flash photolysis; the
amount of T–T absorption ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) s. temperature or surfactant concentration reflects that of the dimer.
s. temperature or surfactant concentration reflects that of the dimer.

2 Experimental
Compounds 1–4 were synthesized by procedures described in ref. 1. Ru(bpy)32+, naphthalene, naphthalene-1-sulfonic acid, tetraphenylphorphine (TPP), fluoresceine and Rose Bengal as well as SDS, CTAB, Triton X-100, acetonitrile (Uvasol), ethanol and heavy water (Merck) were used as commercially available. Water was from a Millipore (Milli Q) system.The steady-state absorption spectra were recorded on spectrophotometers (Hitachi 330, Perkin-Elmer 554 and Hewlett Packard 8453) in 1 cm cuvettes unless otherwise indicated. Generally the concentration of dyes was varied in the 0.3–10 μM range and the pH value in aqueous solution was near neutral. The relative absorbances were measured at the appropriate wavelengths using Arel = (A − A0)/(Amax − A0) for the monomer, where A0 and Amax refer to neat water and a sufficiently high surfactant concentration, respectively. The fluorescence and phosphorescence spectra were recorded on Shimadzu RF5000, Spex-Fluorolog and Perkin-Elmer (LS-5) spectrofluorimeters. The quantum yield Φf was determined using optically matched samples of sodium fluoresceine in aqueous solution (pH 12) as a reference (Φf = 0.92 for λexc = 366 nm and 0.8 for λexc = 436 nm)25 and absorbances of 0.05 at λexc (1 cm pathlength). The experimental error in Φf is typically ± 20%. Higher temperatures were achieved by a thermostat, whereas the cooling of the sample down to − 196°C was performed in a transparent quartz Dewar flask.
The laser set-up, using transient digitizers (Tektronix 7912AD and 390AD), was the same as that used previously.23,24 Most experiments were performed with the 425 nm output of a dye laser pumped by the 308 nm pulse of a XeCl excimer laser (Lambda Physik, EMG 200). Phosphorescence of singlet molecular oxygen at 1269 nm was detected after the pulse using a cooled Ge detector (North Coast, EO 817FP), a silicon and an interference filter and an amplifier (Comlinear, CLC-103) as described elsewhere.26 Unless otherwise indicated, the measurements were carried out at room temperature.
3 Results
3.1 Ground state properties
The thiamonomethinecyanine dyes at low concentration (e.g. 1 μM) at room temperature are mostly present as dimers in aqueous solution and mostly as monomers in ethanol (Table 1).| Compound | Solvent | λ M/ nm | λ D/ nm | K D/ 104 M−1 |
|---|---|---|---|---|
| a In solution at 24°C. b Values in parentheses refer to a shoulder. c J-aggregate. | ||||
| 1 | Ethanol | 440 | ||
| Water | 416(448)b | |||
| 2 | Ethanol | 443 | ||
| Water | 417(448), 480c | 70 | ||
| Heavy water | 485c | |||
| 20% ethanol | 450 | 9 | ||
| 3 | Ethanol | 456 | ||
| Water | 421(466) | |||
| 40% ethanol | 460 | 50 | ||
| 4 | Ethanol | 448 | ||
| Water | 416(457) | |||
| 50% ethanol | 450 | 1 | ||
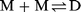
| (1) |
This is illustrated in Fig. 1a for two cases. For 2 the respective maxima are at λD = 417 nm and λM = 443 nm and a weaker peak or shoulder in the absorption spectra at 448 and 420 nm, respectively. On increasing the temperature between 25 and 99°C the relative amount of monomer of 2 (Fig. 2a) or 4 (Fig. 2b) in aqueous solution increases from <5% to nearly 100%. Similar dependences were observed for 1 or 3.
 | ||
| Fig. 1 (a) Absorption spectra of 2 (μM) in aqueous solution (solid line) and ethanol (broken line) and (b) fluorescence emission (right) and excitation spectra (left) in aqueous solution (λf = 521 nm, λexc = 423 nm) at 25°C. | ||
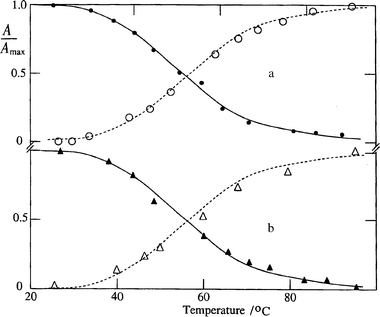 | ||
| Fig. 2
Plots s. temperature of the relative amount of monomer (open symbols) and T–T absorption (closed symbols) after the 425 nm pulse for (a) 2 and (b) 4
in aqueous solution.
| ||
The dimer nature of the band at shorter wavelengths is
further established by the aggregation number of ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 1.6 for 2 in water at 25°C (using concentrations of 0.1–1 μM) and 2.0 ± 0.1 in a mixture with 20% ethanol. Note that the monomer band is larger and better resolved in the presence of ethanol than in neat water and the evaluation of the ratio of absorbances
should favour that of the monomer.21
For 4 in a mixture with 50% ethanol the aggregation number is 2.0 ± 0.03. The dimerization constant was determined from the dependence of the absorption of the monomer band on the total dye concentration per A(λM).23,27KD
= 7 × 105 M−1 for 2 in aqueous solution and is lower in the presence of ethanol as also
in the other cases (Table 1).
1.6 for 2 in water at 25°C (using concentrations of 0.1–1 μM) and 2.0 ± 0.1 in a mixture with 20% ethanol. Note that the monomer band is larger and better resolved in the presence of ethanol than in neat water and the evaluation of the ratio of absorbances
should favour that of the monomer.21
For 4 in a mixture with 50% ethanol the aggregation number is 2.0 ± 0.03. The dimerization constant was determined from the dependence of the absorption of the monomer band on the total dye concentration per A(λM).23,27KD
= 7 × 105 M−1 for 2 in aqueous solution and is lower in the presence of ethanol as also
in the other cases (Table 1).
In heavy water (which was also used for singlet oxygen measurements) the dimer band of 2 is weak and a new band centered at λJ = 485 nm appears (Fig. 3a). We assign this band to a J-aggregate (see Discussion) since the monomer (λM = 440 nm) can be excluded. On increasing the temperature between 25 and 90°C the values of A420 and A440 increase and A485 decreases correspondingly, i.e. the amount of J-aggregate decreases and the contents of both dimer and monomer increase. A J-aggregate was also observed in aqueous solution at concentrations higher than 10 μM and in diluted 2 (1 μM) in the presence of 0.1 M KCl (Table 1 and Fig. 3b). These effects are specific for 2 and were not registered for 1 and 3. Nevertheless, for 4 at larger KCl concentrations of 0.5 M a J-aggregate also appeared.
 | ||
| Fig. 3 Absorption spectra of 2 (1 μM) (a) in heavy water at 25°C (broken line) and 75°C (solid line) and (b) in aqueous solution at 25°C for KCl = 0 M (solid line) and 0.1 M (broken line). | ||
3.2 Luminescence properties
The monomethine dyes show fluorescence; the quantum yield at 24°C is Φf = 0.06–0.08 for 1–4 in aqueous solution and Φf = 0.003–0.006 in ethanol (Table 2). The fluorescence excitation spectra (using concentrations of 0.3-3 μM) resemble those of the absorption with regard to the results that the major maximum originates from the dimer in water and from the monomer in ethanol. In addition, a weaker peak due to the minor amount of monomer in water becomes apparent, as is shown in Fig. 1b for 2. For 1, 3 and 4 in aqueous solution only the most intense peak in the fluorescence excitation spectrum is similar to the absorption spectrum of the dimer, whereas the less intense peak in the fluorescence excitation spectrum is red-shifted with respect to λM, e.g. for 1 by ca. 20 nm. The fluorescence emission spectrum of 2 in water has a maximum at λf = 520 nm and is independent of the wavelength of excitation, λexc = 423 nm (Fig. 1b) or 463 nm. In ethanol λf is blue-shifted by ca. 10 nm and the spectrum is independent of λexc (430 or 455 nm). These findings demonstrate that both the monomer and dimer fluoresce.| Compound | Solvent | λ fex/ nm | λ f/ nm | Φ f | |
|---|---|---|---|---|---|
| a In air-saturated solution at room temperature. b Values in parentheses refer to the minor band. | |||||
| 1 | Ethanol | 423 | 442 | 505 | 0.003 |
| Water | 420 | (457)b | 510 | 0.06 | |
| 2 | Ethanol | 429 | 454 | 509 | 0.003 |
| Water | 426 | (463) | 520 | 0.06 | |
| 3 | Ethanol | (425) | 467 | 522 | 0.006 |
| Water | 422 | (469) | 543 | 0.055 | |
| 4 | Ethanol | (424) | 456 | 512 | 0.004 |
| Water | 422 | (463) | 522 | 0.08 | |
The dyes show phosphorescence with maxima around 570 nm in ethanol at − 196°C. From the onset of the spectrum at 550 nm for 1, 2 and 4 (for 3 the intensity is too weak under our conditions) a triplet energy of ET = 220 kJ mol−1 was obtained. The phosphorescence excitation spectrum has a major maximum originating from the monomer. The phosphorescence decays by first-order kinetics and the lifetime is 0.26–0.29 s. The phosphorescence of the dimer was observed for 1-4 in ethylene glycol–water mixtures (2:1) at − 196°C. The maximum is located at 572 nm, except for 3 where it is shifted to 595 nm. The presence of two methoxy groups should lower the triplet energy, as for other monomethine dyes. Possible reasons are hydrogen bonding to the solvent or intramolecular charge transfer.
Attempts to detect formation of singlet molecular oxygen for 1 in air- or oxygen-saturated D2O at room temperature were successful, albeit the signal is low with respect to TPP in toluene and even lower with respect to Rose Bengal in D2O. Our estimation for the yield of singlet oxygen for 1 in heavy water is 0.08.
3.3 Triplet state absorption properties
Upon pulsed excitation at 425 nm the transient difference spectrum of 1–4 (3–30 μM) in aqueous solution exhibits a bleaching with a maximum (λbl) around 420 nm, an absorption maximum (λTT) at 450 nm and a minor broad band at 500–700 nm. An example is shown in Fig. 4a for 2. The decay is unimolecular and reflects only the recovery of the initial absorption. The transient appears during the pulse and is quenched by oxygen, the rate constant is kO2 = (1.4–2.5) × 109 M−1 s−1 (Table 3). The transient is therefore attributed to the triplet state. Similar spectra were recorded upon excitation at 308 nm and with μs flash photolysis. | ||
| Fig. 4
Difference absorption spectra in argon-saturated aqueous solution at 25°C of (a) 2 (10 μM, pH 7) at 0.1 (○), 50 (△) and 200 μs (●) after the 425 nm pulse; broken line: ground state absorption and (b) of 1 (pH 2.6) at <1 (○) and 10 μs (●) with naphthalene-1-sulfonic acid (A3083) as sensitizer.
| ||
| Compound | λ bl/nm | λ TT b/nm | τ T c/ms | k O2/109 M−1 s−1 |
|---|---|---|---|---|
| a In aqueous solution at 25°C. b Minor band at 500–700 nm. c Under argon for dye concentrations of 1 μM using μs flash photolysis. d Values in parentheses refer to λexc = 425 nm. | ||||
| 1 | 420 | 450 | <0.2 (0.10)d | 2.5 |
| 2 | 420 | 440 | 0.31 (0.15) | 2.4 |
| 3 | 420 | 460 | 0.48 (0.33) | 1.4 |
| 4 | 420 | 460 | 0.91 (0.18) | 2.0 |
Only a weak transient absorption and bleaching could be detected upon direct excitation of 1–4 in ethanol or other polar organic solvents, e.g. acetonitrile. Thus we attribute the observed triplet state in aqueous solution to that of the dimer. This is supported by the result that with increasing temperature the amount of triplet absorption or bleaching decreases (Fig. 2). The observed bleaching (Fig. 4a) is mainly due to population of the less absorbing triplet state. This follows from the result that the rate constant for depletion of the dimer ground state matches that of triplet decay either in the absence or presence of oxygen.
The triplet lifetime (τT) of the dimer of 1–4 in argon-saturated aqueous solution is in the 0.1–0.9 ms range (Table 3). Note that triplet decay is limited by self-quenching and that the triplet lifetime is longer when μs flash photolysis is used. This is a consequence of the lower dye concentration used (with respect to the laser experiments), thereby reducing self-quenching, and partly to the lower triplet concentration, thus avoiding T–T annihilation. This general behaviour has also been reported for related cases.23,28
The quantum yield of intersystem crossing of the monomeric
monomethine dyes is very low, ca. 10−3. A substantial
increase in Φisc was found for the dimers. As a reference for direct excitation at 425 nm, we used Ru(bpy)32+ in aqueous solution (Φisc
= 1); its molar absorption coefficient is ε370
= 1.8 × 104 M−1 cm−1.29
From the transient absorption of Ru(bpy)32+s. laser intensity and the signal of 2 using ε420
= 1.0 × 105 M−1 cm−1 (see below) under optically matched conditions, the value Φisc
= 0.2 ± 0.05 was estimated. Only slightly lower values were estimated for the three other dyes.
3.4 Energy transfer
Upon 308 nm excitation of naphthalene (A308 = 3) in argon-saturated ethanol in the presence of 2, the difference spectra show, apart from the T–T absorption band of the sensitizer (energy donor) centered at 415 nm, a bleaching around 455 nm and a weak band in the red spectral range above 500 nm (not shown). Decay of the donor triplet at 415 nm matches the increase of the acceptor triplet at 455 nm (e.g. for 50% quenching). The rate constant for energy transfer is kET>1 × 1010 M−1 s−1 and was determined from the linear dependence of the first-order decay kinetics of the donor triplets. acceptor dye concentration.
On the other hand, upon excitation of naphthalene-1-sulfonic acid (A3083) in argon-saturated aqueous solution (pH 2.6–3) in the presence of 1, 3 or 4, the difference spectra show bleaching at 420 nm, a major absorption peak at 450 nm and a weak band in the red spectral range (Fig. 4b). Decay of the triplet donor also matches the formation of the acceptor triplet.
Based on a molar absorption coefficient of ε440
= 1.5 × 105 M−1
cm−1 for the donor triplet30 and corrected for 100% quenching, that of the acceptor triplet in the case of 1 is ε450
= 1 × 105 M−1
cm−1. Note that the amount of directly generated triplet state of 1 is negligibly small under the applied conditions. These findings support the triplet nature of the observed
transients under direct excitation of the thiamonomethinecyanine dyes and their assignment as originating from the monomer and dimer in ethanol and aqueous solution, respectively.
3.5 Effects of surfactants
On addition of CTAB (<0.1 mM) as cationic surfactant to 2 in aqueous solution, the dimer band decreases and a new band centered at 485 nm appears (Fig. 5) which we assign to a J-aggregate. This is in agreement with the results for 2 in the absence of surfactants described above (Fig. 3). The plot of A420s. the CTAB concentration decreases and A485 increases
correspondingly (Fig. 6). The CTAB concentration for the 50% values, [CTAB]1/2 is ca. 20 μM. At [CTAB] higher than the critical micellization concentration (c.m.c., 0.92 mM)31 the monomer band becomes more pronounced and that of the J-aggregate much less intense (Fig. 5). In contrast to 2, no
specific J-aggregate was observed for 1, 3
and 4. In these cases the dimer band decreases in the concentration range 0–0.1 mM and the monomer band increases at a [CTAB] larger than
the c.m.c.
![Absorption spectra of 2 at [CTAB] (a) 0, (b) 0.01, (c) 0.02, (d) 1.0 and (e) 8.0 mM in aqueous solution at 25°C.](/image/article/2001/CP/b005683i/b005683i-f5.gif) | ||
| Fig. 5 Absorption spectra of 2 at [CTAB] (a) 0, (b) 0.01, (c) 0.02, (d) 1.0 and (e) 8.0 mM in aqueous solution at 25°C. | ||
 | ||
| Fig. 6
Plots of A420 (●), A450 (○), A485 (♦) and −
ΔA420 (△) s. the CTAB concentration for 2 in aqueous solution at 25°C.
| ||
The presence of SDS as anionic surfactant does not lead to
a J-aggregate for 2, but the monomer band increases s. [SDS] and the dimer band decreases at [SDS]1/2 of ca. 10 mM which almost coincides with the c.m.c. In contrast, Triton X-100 as
non-ionic surfactant does not lead to marked changes in the absorption spectra of 1–3 around the c.m.c. of 0.26 mM31
and the [surfactant]1/2 values are shifted to much higher concentrations of 1–8 mM.
The fluorescence intensity (If) of 1 or 3 does not change
drastically with [surfactant], but the excitation spectrum is due to the monomer, when [CTAB] or [SDS] is above the c.m.c.
or when [Triton X-100] is even much higher. The transient difference spectra of 1–4 in the presence of surfactants (<5 μM), upon pulsed excitation at 425 nm, are not changed; they have a bleaching at 420 nm, an absorption maximum at
λTT and a minor broad band at 500–700 nm. The amount of T–T absorption decreases s. CTAB concentration with [CTAB]1/2 being in the
10–20 μM range (Fig. 6 and Table 4).
| Compound | Additive | λ M/ nm | λ J/ nm | [Surfactant]1/2/ mM |
|---|---|---|---|---|
| a In aqueous solution at 25°C; the c.m.c. = 8.1, 0.92 and 0.26 mM for SDS, CTAB and Triton X-100, respectively. b Values in parentheses refer to T–T absorption. | ||||
| 1 | CTAB | 445 | 0.01 (0.01)b | |
| 2 | KCl (0.1M) | None | 480 | |
| CTAB | 443 | 480 | 0.02 | |
| Triton X-100 | 450 | 8 | ||
| SDS | 450 | 10 | ||
| 3 | CTAB | 460 | 0.03 | |
| 4 | CTAB | 470 | 0.03 | |
| KCl (0.5 M) |
None | 480 | ||
4 Discussion
4.1 Thiamonomethinecyanine dyes in homogeneous media
Self-association of dyes in solution can be caused by van der Waals forces, hydrophobic interaction and hydrogen bonding either intermolecular or with the solvent.16,32 Formation of dimers in water, due to its high relative permittivity, can be facilitated by the reduction of repulsion between similarly charged ions. In contrast, the dyes in ethanol are present as monomers because of the lower relative permittivity. Moreover, dimer formation reduces hydrophobic interactions with water. Hydrogen bonding with water molecules is the main driving force for dimerization of xanthene dyes, where the enthalpy, ΔHD, and entropy, ΔSD, dimerization values are negative.32 For 1 the thermodynamic parameters of dimerization in aqueous methanol at 24°C have been reported, ΔHD = − 54 kJ mol−1 and ΔSD = − 97 J mol−1 K−1.21 We suggest that for 2–4 hydrogen bonding is also the main driving force for dimerization.Dimerization of 1–4 is characterized by an intense band at shorter wavelengths and a weak band at longer wavelengths in aqueous solution (Fig. 1a and Table 1), the effects of temperature (Fig. 2) and concentration and an aggregation number of 2. Two bands in the absorption spectra of dimers originate from a splitting of the excited singlet energy level into levels of higher and lower energy. According to the Kasha exciton theory33 for a parallel orientation of monomers in a dimer (face-to-face) or their head-to-tail alignment, one absorption band is expected which is blue- or red-shifted with respect to that of the monomer. For dimers of the four monomethine dyes examined, however, two bands in both absorption and fluorescence excitation spectra were observed (Fig. 1 and Table 1). Therefore, in our case the face-to-face and head-to-tail alignments are excluded and an oblique structure remains. Assuming that the two monomer dyes stack on top of each other, the angle between the polarization axis of the monomers in the dimer was estimated to be 50–60°. The distance between the centers of gravity of the two molecules was estimated as 0.9–1.0 nm. This is consistent with the distance for thiacarbocyanine dimers in aqueous solution (0.85–0.94 nm)23 and rhodamine 6G in ethanol (1.1 nm).3
The larger KD of 3 with respect to 4 might be a result of the presence of a second OCH3 group; this, in addition to sulfur and nitrogen atoms, might account for the additional hydrogen bonding. The J-aggregates are generally characterized by their rather sharp long-wavelength peak.1,16,34 The observed J-aggregate for the specific case of 2 in heavy water (Fig. 3a) and in aqueous solution in the presence of KCl (Fig. 3b) may be due to the chlorine atom.16,35 The J-aggregates of 2 were shown to be formed from dimers.6
4.2 Excited singlet state behaviour
The dimers of 1–4 exhibit fluorescence (Fig. 1b) with a moderate yield being 10–20 times larger than the Φf value of the monomers (Table 2). The lower Φf and the negligible Φisc values of monomers with respect to their dimers indicate efficient internal conversion, caused by rotation of the naphthothiazolium residue around the monomethine chain.Fluorescence in low yield has been reported for dimer molecules of Acridine Orange8,9 and rhodamine 6G (Φf⩽0.007).36 On the other hand, dimers of squaraine,37 Zn–phthalocyanine,38 certain xanthene dyes39 and thiacarbocyanines23 do not exhibit fluorescence at ambient temperature. This difference might follow from different orientations of the molecules in a dimer; i.e. an alignment other than face-to-face may be the reason for the fluorescence of the dimeric monomethine dyes. This is also confirmed by the observation of two maxima in fluorescence excitation spectra for dimers (Fig. 1b) which correspond to two peaks in absorption spectra for 0.2–0.5 μM monomethines. The estimation of the angle between the transition moments of the monomers in the dimer as well as the monomer–monomer distances appeared to be in a good agreement with those obtained from both absorption and excitation fluorescence spectra.
4.3 Triplet state properties
Assignment of the observed transient of 1–4 in aqueous solution to the triplet state follows from its formation within the pulse width, the effect of oxygen on the decay and a comparison of the spectra and λTT under direct and sensitized excitation conditions (Fig. 4 and Table 3). The decrease in the amount of triplet state with increasing temperature (Fig. 2) further demonstrates that the observed triplet state originates from the excited dimer. The triplet properties of dimers have been reported for Methylene Blue,7 Acridine Orange,8,9 Rose Bengal,10 triarylmethane dyes,11 triarylpyrylium salts,12 phthalocyanines,13 porphines14 and thiacarbocyanines.23Formation of the monomer by photodissociation of the dimer for eosin40 and thermal deagreggation upon heating by the laser pulse for pseudoisocyanine and thiacarbocyanines19 has been reported. However, this is not the case for the monomeric thiamonomethinecyanine dyes since the transient is the triplet state. The substantial increase in Φisc on replacing ethanol by water is a consequence of the presence of dimers (Scheme 1). Generally, singlet–triplet splitting in dimers is smaller than in monomers.33 In addition, rotation of the naphthothiazolium residue around the monomethine chain should be more restricted in dimers than in monomers. In glassy media at − 196°C, where phosphorescence of the monomers and dimers was recorded, the hindrance of rotation is probably even stronger.
 | ||
| Scheme 1 | ||
4.4 The role of surfactants
Micelles have been investigated for various dyes.30,41,42 It is well-known that dyes can deaggregate in the presence of surfactants.16,24,43,44 On the other hand, surfactants can influence the aggregation of dyes and ion-pairing and were shown to be a pre-requisite for this process.45 Thus a knowledge of the aggregation–deaggregation process is important. Dye molecules in microheterogeneous media reveal a strong enhancement of Φf, e.g. for cyanine dyes,46,47 and a higher photostability, e.g. for Rose Bengal,48 Methyl Orange49 or merocyanine 540.50 Premicellar aggregation has been reported for CTAB and anionic dyes.49For the cationic CTAB one might expect that the hydrophobic
interaction between the monomer and the surfactant,
which causes the deaggregation, would be enhanced by the
attracting Coulombic forces. The new narrow band of 2 in the
[CTAB] range 10–100 μM (Fig. 5) is assigned to J-aggregates. In the c.m.c. region and above, the J-aggregates are converted into monomers due to association of the dye with the micelle.
Thus the 2/CTAB
system is characterized by three regimes which are separated by the formation of ion pairs, formation of J-aggregates and their conversion into solubilized monomers with increasing [CTAB]. For the three other dyes, where J-aggregates are not formed, the [CTAB]1/2 values in Table 4 refer to dimer splitting. For the 2/CTAB system formation of solubilized monomers was also observed by T–T absorption; the signals decrease s. CTAB concentration with [CTAB]1/2
= 10 μM (Fig. 6 and Table 4).
The conversion into solubilized monomers with increasing [CTAB] is reflected by the fluorescence excitation spectra of 1–3, indicating formation of monomers at [CTAB] higher than the c.m.c. This could also be observed for the other two surfactants. Concerning the question as to the origin of the relatively small changes of If, two counterbalancing effects have to be considered. One is the lower Φf value of the monomer, at least in ethanol (Table 2), and the other effect is due to the properties of micelles, e.g. an enhancement of If due to a larger microviscosity above the c.m.c.31,51 These opposing effects contribute in almost all cases, i.e. CTAB, SDS or Triton X-100.
5 Conclusions
In aqueous solution dimers of 1–4 are formed. The major deactivation pathway of the excited state of the monomeric thiamonomethinecyanines is internal conversion, whereas dimerization results in both strong enhancement of intersystem crossing and moderate increase of fluorescence. Ionic surfactants promote the splitting of dimers into solubilized monomers and the formation of J-aggregates for the 2/CTAB system in the premicellar region.Acknowledgements
We thank Professor Kurt Schaffner for his support, Professor B. I. Shapiro for providing the four thiamonomethinecyanine dyes and Mr. Leslie J. Currell for technical assistance. A.K.C. is grateful to the Deutsche Forschungsgemeinschaft and the Russian Fund of Basic Researches (00-03-32300) for financial support.References
- The Theory of the Photographic Processes, ed. T. H. James, Macmillan, New York, 4th edn., 1977. Search PubMed.
- B. Kopainsky, J. K. Hallermeier and W. Kaiser, Chem. Phys. Lett., 1981, 83, 498 CrossRef CAS; B. Kopainsky, J. K. Hallermeier and W. Kaiser, Chem. Phys. Lett., 1982, 87, 7 CrossRef CAS.
-
V. I. Yuzhakov, Usp. Khim., 1992, 61, 1114 (Engl. transl. in Russ. Chem. Re
![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ., 1992, 61, 613). Search PubMed.
., 1992, 61, 613). Search PubMed. - A. K. Chibisov and T. D. Slavnova, J. Photochem., 1978, 8, 285 Search PubMed.
- M. Mauerer, A. Penzkofer and J. Zweck, J. Photochem. Photobiol. B, 1998, 47, 68 Search PubMed.
- V. I. Avdeeva and B. I. Shapiro, Zh. Nauchn. Prikl. Fotogr., 1999, 44, 20 (Engl. transl. in Sci. Appl. Photogr., 1999, 41, 129). Search PubMed.
- R. M. Danzinger, K. H. Bar-Eli and K. Weiss, J. Phys. Chem., 1967, 71, 2633 CrossRef.
- R. W. Chambers and D. R. Kearns, J. Phys. Chem., 1968, 72, 4718 CrossRef CAS.
- F. Wilkinson, D. R. Worrall and L. F. Vieira Ferraira, Spectrochim. Acta Part A, 1992, 48, 135 CrossRef.
- Y. Chen, T. Urano, T. Karatsu, S. Takahara, T. Yamaoka and K. Tokumaru, J. Chem. Soc., Perkin Trans. 2, 1998, 2233 RSC.
- G. Jones, C. Oh and K. Goswami, J. Photochem. Photobiol. Chem. A, 1991, 57, 65 Search PubMed.
- I. Lampre, D. Markovitsi, A. Sharonov and M. Veber, Chem. Phys. Lett., 1998, 293, 423 CrossRef.
- A. Ferencz, D. Neher, M. Schulze, G. Wegner, L. Viaene and F. C. De Schryver, Chem. Phys. Lett., 1995, 245, 23 CrossRef CAS.
- H. Levanon, A. Regev and K. P. Das, J. Phys. Chem., 1987, 91, 14 CrossRef CAS; P. Jaegermann, M. Plato, B. von Malzan and K. Möbius, Mol. Phys., 1993, 78, 1057 CAS; M. Hugerat, A. van der Est, E. Ojadi, L. Biczok, H. Linschitz, H. Levanon and D. Stehlik, J. Phys. Chem., 1996, 100, 495 CrossRef CAS; M. Asano-Someda, T. Ichino and Y. Kaizu, J. Phys. Chem. A, 1997, 101, 4484 CrossRef CAS.
- J. K. Hicks and P. J. Hore, Chem. Phys. Lett., 1995, 237, 183 CrossRef CAS.
- A. H. Herz, Photogr. Sci. Eng., 1974, 18, 323 Search PubMed.
- B. Armitage, J. Retterer and D. F. O'Brien, J. Am. Chem. Soc., 1993, 115, 10786 CrossRef CAS.
- V. Sundström and T. Gillbro, J. Chem. Phys., 1985, 83, 2733 CrossRef.
- R. F. Khairutdinov and N. Serpone, J. Phys. Chem. B, 1997, 101, 2602 CrossRef CAS.
- J. Moll, S. Daehne, J. R. Durrant and D. A. Wiersma, J. Chem. Phys., 1995, 102, 6362 CrossRef CAS; K. Minoshima, M. Taiji, K. Misawa and T. Kobayashi, Chem. Phys. Lett., 1994, 218, 67 CrossRef CAS.
- Z. Zhang, J. Hao, B. Wu and H. Yuan, J. Imag. Sci. Technol., 1995, 39, 373 Search PubMed.
- V. I. Avdeeva and B. I. Shapiro, Zh. Nauchn. Prikl. Fotogr., 1997, 42, 27 (Engl. transl. in Sci. Appl. Photogr., 1998, 39, 543). Search PubMed.
- A. K.Chibisov, G. V. Zakharova and H. Görner, Phys. Chem. Chem. Phys., 1999, 1, 1455 RSC.
- A. K. Chibisov, V. I. Prokhorenko and H. Görner, Chem. Phys., 1999, 250, 47 CrossRef CAS.
- C. A. Parker, Photoluminescence of Solutions, Elsevier, New York, 1977. Search PubMed.
- H. Görner, Chem. Phys. Lett., 1998, 282, 381 CrossRef CAS.
- T. Matsubara and T. Tanaka, J. Imag. Sci., 1991, 35, 274 Search PubMed.
- A. K. Chibisov, G. Zakharova, H. Görner, Yu. A. Sogulyaev, I. L. Mushkalo and A. I. Tolmachev, J. Phys. Chem., 1995, 99, 886 CrossRef CAS.
- G. Ponterini and F. Momicchioli, Chem. Phys., 1991, 151, 111 CrossRef CAS; N. Nakashima and T. Kunitake, J. Am. Chem. Soc., 1982, 104, 4261 CrossRef CAS.
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1.
- J. C. Scaiano, CRC Handbook of Organic Photochemistry, CRC Press, Boca Raton, FL, 1989, vol. 2 Search PubMed; K. Kalyanasundram, Photochemistry in Microheterogeneous Systems, Academic Press, New York, 1987. Search PubMed.
- O. Valdes-Aguilera and D. C. Neckers, Acc. Chem. Res., 1989, 22, 171 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Molecular Spectroscopy, Butterworth, London, 1965, p. 371 Search PubMed; E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721 Search PubMed; E. G. McRae and M. Kasha, in Physical Processes in Radiation Biology, ed. L. Augenstein, R. Mason and B. Rosenberg, Academic Press, New York, 1964, p. 23. CrossRef CAS.
- W. West, in Dye Sensitization, ed. W. F. Berg, U. Mazzucato, H. Meier and G. Semerano, Focal Press, London, 1970, p. 105. Search PubMed.
-
B. I. Shapiro, Usp. Khim., 1994, 63, 243 (Engl. transl. in Russ. Chem. Re., 1994, 63, 231). Search PubMed.
- P. Bojarski, A. Matczuk, C. Bojarski, A. Kawski, B. Kuklinski, G. Zurkowska and H. Diehl, Chem. Phys., 1996, 210, 485 CrossRef CAS.
- H. Chen, M. S. Farahat, K.-Y. Law and D. G. Whitten, J. Am. Chem. Soc., 1996, 118, 2584 CrossRef CAS.
- R. M. Negri, A. Zalts, E. A. San Roman, P. F. Aramendia and S. E. Braslavsky, Photochem. Photobiol., 1991, 53, 317 Search PubMed.
- K. Kemnitz and K. Yoshihara, J. Phys. Chem., 1991, 95, 6095 CrossRef CAS.
- R. Dunsbach and R. Schmidt, J. Photochem. Photobiol. Chem. A, 1995, 85, 275 Search PubMed.
- F. M. Menger, Acc. Chem. Res., 1979, 12, 111 CrossRef CAS; J. K. Thomas, Chem. Re., 1980, 80, 283 Search PubMed.
- M. H. Gehlen and F. C. De Schryver, Chem. Re., 1993, 93, 199 Search PubMed.
-
V. I. Yuzhakov, Usp. Khim., 1992, 61, 1114 (Engl. transl. in Russ. Chem. Re., 1992, 61, 613). Search PubMed.
- M. Fischer and J. Georges, Spectrochim. Acta, 1997, A53, 1419 Search PubMed.
- T. Sato, Y. Yonezawa and H. Hada, J. Phys. Chem., 1989, 93, 14 CrossRef CAS; P. Bilski, R. N. Holt and C. F. Chignell, J. Photochem. Photobiol. Chem. A, 1997, 110, 67 Search PubMed; P. Pal, H. Zeng, G. Durocher, D. Girard, R. Giasson, L. Blanchard, L. Gaboury and L. Villeneuve, J. Photochem. Photobiol. Chem. A, 1996, 98, 65 Search PubMed.
- R. Humphry-Baker, M. Grätzel and R. Steiner, J. Am. Chem. Soc., 1980, 102, 847 CrossRef CAS.
- N. Nakashima and T. Kunitake, J. Am. Chem. Soc., 1982, 104, 4261 CrossRef CAS.
- P. Bilski and C. F. Chignell, J. Photochem. Photobiol. Chem. A, 1994, 77, 49 Search PubMed.
- M. H. Gehlen, M. Ferreira and M. G. Neumann, J. Photochem. Photobiol. Chem. A, 1995, 87, 55 Search PubMed.
- N. S. Dixit and R. A. Mackey, J. Am. Chem. Soc., 1983, 105, 2928 CrossRef CAS.
- A. Datta, D. Mandal, S. K. Pal and K. Bhattacharyya, Chem. Phys. Lett., 1997, 278, 77 CrossRef CAS; F. Ortica and G. Favaro, J. Lumin., 1996, 68, 137 CrossRef CAS.
| This journal is © the Owner Societies 2001 |