Sub-ppt detection limits for copper ions with Gly-Gly-His modified electrodes†
Wenrong Yang, David Jaramillo, J. Justin Gooding*, D. Brynn Hibbert, Rui Zhang, Gary D. Willett and Keith J. Fisher
School of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia.. E-mail: justin.gooding@unsw.edu.au
First published on 18th September 2001
Abstract
An electrochemical metal ion sensor has been developed with a detection limit of less than 0.2 ppt by the covalent attachment of the tripeptide Gly-Gly-His as a recognition element to a 3-mercaptopropionic acid modified gold electrode.
Detecting a number of metal ions at very low concentrations directly in the field is one of the key targets of environmental chemists. Electrochemical sensors have the potential to allow multi-ion analysis in the field because of their small size, low power requirements and the need for little or no sample pretreatment. The majority of research into chemically modified electrodes for the detection of metal ions involves the synthesis and testing of macrocyclic ligands to provide the sensing interface with selectivity for a given metal ion and the ability to pre-concentrate the metal. In nature however, binding metals is achieved with a high degree of selectivity using peptide motifs rather than macrocyclic ligands. Considering the development of biosensors that exploit Nature’s methods of selective recognition and the extensive body of literature on the complexation of metal ions with peptides,1,2 it is surprising how little research has been conducted into the development of solid state metal ion sensors based on peptide ligands.3 Here we describe an electrode modified with the oligopeptide Gly-Gly-His with an extraordinarily low detection limit in the sub ppt range (pM) for copper ions.
Electrodes modified with Gly-Gly-His were prepared using polycrystalline gold electrodes cleaned as described previously.4,5 Self-assembly of 3-mercaptopropionic acid (MPA) was achieved by soaking the electrodes in a 1.0 mM MPA solution in ethanol for 12 hours. The carboxylic acid terminated self assembled monolayer (SAM) was activated using a mixture of 15 mM N-hydroxysuccinimide (NHS) and 75 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and 2-(N-morpholino)ethanesulfonic acid (MES) buffer solution (pH 5.5) for 30 minutes.6 The resultant succinimide ester monolayers were reacted for 30 minutes in a solution of Gly-Gly-His (50 mg ml−1) in MES buffer to give the modified electrode shown in Fig. 1 where the tripeptide is attached through the amino group of the first glycine and terminates with the carboxylic acid of the histidine.
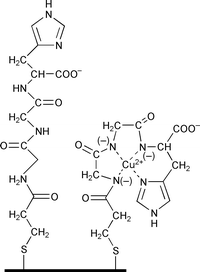 | ||
| Fig. 1 Schematic of the Gly-Gly-His modified electrode with and without complexed Cu2+. | ||
Copper was accumulated at the Gly-Gly-His modified electrode (GGHME) in an open circuit potential for 10 minutes in a 0.05 M ammonium acetate buffer solution (pH 7.0) containing copper nitrate. Cyclic voltammetry (using a BAS 100B, Bioanalytical System Inc., USA) measured after removal of the electrode from the copper nitrate solution, rinsing and then placing in a copper free ammonia acetate buffer solution showed reversible electrochemistry with a half-wave potential at −55 mV versus Ag|AgCl|3.0 M KCl which was attributed to the half cell Au|Cu(II)complex, Cu(I)complex as reported previously for cysteine modified electrodes.7–9 In the absence of accumulated copper no such electrochemistry was observed. Copper free electrodes could be regenerated by elimination of Cu(II) by holding the working electrode at +0.5 V for 30 seconds in 0.1 M HClO4.
That Cu(II) is tightly bound to the GGHME is demonstrated by no diminution of the peak currents with multiple scans, despite the absence of any copper in the test solution. Furthermore, the peak current varies linearly with scan rate, behaviour indicative of the redox active species being strongly bound to the electrode.10 Hence we propose the copper is bound to the GGHME as shown in Fig. 1 to give the tetragonal coordination preferred by Cu(II). Evidence for the binding of the copper through the nitrogen on the histidine ring, to give coordination by four nitrogen atoms, was obtained from studies of collision-induced dissociation (CID) using electrospray ionization Fourier transform ion cyclotron resonance mass spectroscopy (FT-ICR MS).† In the absence of copper, the negative ion mass spectrum of Gly-Gly-His in pure methanol showed the pseudo-parent ion [Gly-Gly-His − H] at m/z 268.11 Da and the most prominent fragment ion at m/z 154.06 Da which arises from the cleavage of histidine to leave [Gly-Gly]. However, in the presence of Cu(II) the cleavage of histidine is prevented by its role in the complexing of copper. The pseudo-parent ion [Gly-Gly-His + Cu2+ − 3H] is observed at m/z 331.02 Da with the most common fragment ion at m/z 287.03 Da which corresponds to decarboxylation from the histidine; thus confirming in solution that the binding of the copper is through a deprotonated amine of the peptide bond between Gly and His. These results are consistent with mass spectroscopy results reported previously.11
Osteryoung square wave voltammetry (OSWV) was employed with a GGHME for detection of low levels of copper in environmental samples. This technique is more sensitive than cyclic voltammetry.12Fig. 2a shows OSWV peaks of a GGHME in 0.05 M ammonium acetate buffer solution (pH 7.0) for a series of copper standards adsorbed for ten minutes from solutions with concentrations in the picomolar range, before transfer to the stripping electrolyte. The 3 pM (0.2 ppt) standard could easily be discriminated from the background in which no Cu(II) was added to the buffer (Fig. 2a). The implied detection limit (i.e. <3 pM) is several orders of magnitude below that which we have reported previously for cysteine (1.5 ppb)9 and polyaspartate (0.2 ppb)13 modified electrodes. The low detection limit and strong binding of copper reflects the well known affinity for Gly-Gly-His, the so called copper binding peptide, for Cu(II).1,2
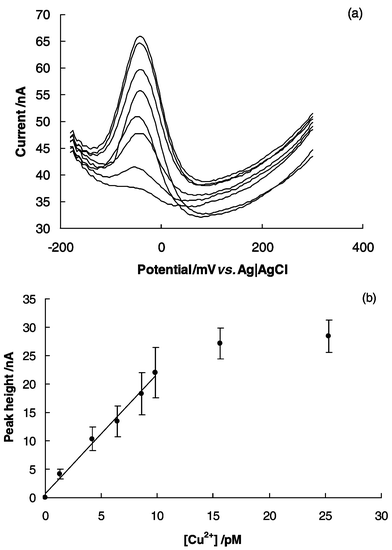 | ||
| Fig. 2 Response of the Gly-Gly-His modified electrode to complexed Cu(II) (a) Osteryoung square wave voltammetry peaks showing an increase in current with copper concentrations of 0, 1.28, 4.2, 6.4, 8.6, 9.8, 15.6, 25.3 pM and (b) calibration curve derived from the OSWV peaks with error bars as ±1 standard deviation of four electrodes prepared on different days. In all cases Cu(II) was accumulated at the Gly-Gly-His modified electrode in an open circuit over 10 minutes in a 0.05 M ammonium acetate buffer solution (pH 7.0) containing copper nitrate, removed, rinsed and then placed in a copper-free ammonium acetate buffer solution. | ||
To the best of our knowledge this detection limit is well below any other reported in the literature [50 pM using the subtractive mode of anodic stripping voltammetry14 (ASV)] and is also well below that observed for most metal ions using anodic stripping voltammetry or catalytic ASV.15 A fluorimetric method has achieved 125 pM16 and electrothermal atomic adsorption spectrometry, where the sample was accumulated via sorption in a knotted reactor, detected 100 pM.17
The calibration curve (Fig. 2b) shows a linear range up to 10 pM with a slope of 2.1 (standard deviation = 0.1) nA pM−1. The standard error in a measurement of concentration made in the middle of the calibration range was 0.33 pM. The electrodes could be regenerated up to 30 times without any apparent change in performance. Four electrodes fabricated on different days with different batches of reagents gave results with RSDs between 10 and 20%, as indicated by the error bars on points in Fig. 2b. The low saturation concentration again reflects the high affinity of the peptide for copper. The selectivity of GGHMEs to copper was tested by measuring the response of the electrode to a number of metals ions commonly found in waste water, shown in Table S1 in the supplementary information. The case of Ni(II) addition of 10 pM of Ni(II) to a solution of 9.8 pM of Cu(II) resulted in a 6.5% decrease in current. Apart from Ni(II) however, none of the other metal ions showed significant interference with the Cu(II) measurement. The interference of Ni(II) is not unexpected considering Ni(II) is also known to form a 4N complex with Gly-Gly-His in solution.2 Analysis of a local tap water was performed by diluting the sample to bring the copper concentration to within the range of the Gly-Gly-His modified electrode. The measured concentration of free Cu(II) in the tap water, when dilutions were accounted for, was 0.27 ppm (standard deviation = 0.03 for 3 samples (i.e.n = 3)) which agreed well with the total copper concentration measured using inductively coupled plasma optical emission spectroscopy (ICP-OES) of 0.33 ppm.
These initial results illustrate the potential for peptide-modified electrodes to be used as metal ion sensors with extraordinarily low detection limits. This work is part of our aim to fabricate a multiple metal ion sensor array, based on peptide modified electrodes, where each electrode in the array gives a different response for a given metal ion to produce a characteristic response pattern for different metal ions. Future work involves exploring other peptide modified electrodes for selectivity for other metal ions.
Notes and references
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385 CrossRef CAS.
- H. Kozlowski, W. Bal, M. Dyba and T. Kowalik-Jankowska, Coord. Chem. Rev., 1999, 184, 319 CrossRef CAS.
- J. J. Gooding, D. B. Hibbert and W. Yang, Sensors, 2001, 1, 75 Search PubMed.
- J. J. Gooding, P. Erokhin and D. B. Hibbert, Biosensors Bioelectronics, 2000, 15, 229 CrossRef CAS.
- Due to the very low concentrations of analyte special care was taken to try to eliminate any copper in solutions or glassware. All solutions were prepared using Milli-Q water and all glassware was soaked in 6 M HNO3 overnight and rinsed thoroughly in Milli-Q water. All OSWV current measurements were recorded relative to a background containing all species except the copper standard..
- J. J. Gooding, V. Praig and E. A. H. Hall, Anal. Chem., 1998, 70, 2396 CrossRef CAS.
- D. W. M. Arrigan and L. Le Bihan, Analyst, 1999, 124, 1645 RSC.
- A. C. Liu, D. C. Chen, C. C. Lin, H. H. Chou and C. H. Chen, Anal. Chem., 1999, 71, 1549 CrossRef CAS.
- W. Yang, J. J. Gooding and D. B. Hibbert, J. Electroanal. Chem., 2001, in the press. Search PubMed.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19 CrossRef CAS.
- P. Hu and J. A. Loo, J. Am. Chem. Soc., 1995, 117, 11314 CrossRef CAS.
- I. Turyan and D. Mandler, Nature, 1993, 362, 703 CrossRef CAS.
- W. Yang, J. J. Gooding and D. B. Hibbert, Analyst, 2001, 126, 1573 RSC.
- Y. Bonfil, M. Brand and E. Kirowa-Eisner, Anal. Chim. Acta, 2000, 424, 65 CrossRef CAS.
- M. Z. Czae and J. Wang, Talanta, 1999, 50, 921 CrossRef CAS.
- K. Watanabe, K. Ohba, A. Iburaim, M. Itagaki and N. Koura, Bunseki Kagaku, 1998, 47, 179 Search PubMed.
- E. Ivanova, K. Benkhedda and F. Adams, J. Anal. At. Spectrom., 1998, 13, 527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: mass spectra and Table S1. See http://www.rsc.org/suppdata/cc/b1/b106730n/ |
| This journal is © The Royal Society of Chemistry 2001 |