Structure and polymorphism of the 10.10.10 Simmons and Park cryptand†
Andrei S.
Batsanov
a,
Alexander J.
Blake
b,
Nicholas M.
Hext
b and
Mark
Mascal‡
*b
aDepartment of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE
bDepartment of Chemistry, University of Nottingham, Nottingham, UK NG7 2RD
First published on 19th December 2000
Abstract
The first crystal structure of a 10.10.10 cryptand has been determined, during which an unusual single crystal phase transition was observed; at room temperature, crystals of macrobicycle 1d are monoclinic (α) but undergo a first-order topotactic phase change at 207 K to a triclinic (β) lattice; full structure determinations at 150 and 210 K were carried out and show that both phases adopt the in–in cryptand conformation.
The foundations of modern host–guest chemistry were laid in two papers published by independent research groups in 1967 and 1968. The former was the first report of alkali metal complexes of a polyether macrocycle,1 and the latter the first report of guest inclusion in a diaza macrobicycle.2 The first result gave birth to the crown ethers, the second to the cryptands, both ‘household’ words in the field of supramolecular chemistry.3
We became interested in the all-hydrocarbon bridged cryptands 1 originally described by Simmons and Park2 from the point of view of isolating the effects of the two N (or HN+) sites to produce linear inclusion complexes with various neutral and ionic guests. Although the ‘original’ crown ether, dibenzo 18-crown-6, has itself been the subject of no less than ninety X-ray crystal structures,4 only one crystallographic study has been published on an ‘original’ cryptand, i.e.1c.5 In this work, the bis(hydrochloride) cryptate [H2(1c)Cl]+ Cl− co-crystallizes with the unusual polyaqua cation H13O6+, the latter species being the main focus of the paper. However, no satisfactory representation of the cage itself was derived from the data, owing to the presence of extensive disorder.
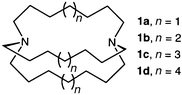
Taking into account the respective melting points in the 1 series indicates that the n.n.n cryptands are conformationally mobile where n is odd, while those where n is even will be conformationally stable.6 This may explain the difficulties the previous workers experienced with 1c. We thus prepared the 10.10.10 cryptand 1d7 and undertook first to obtain an X-ray crystal structure of the empty, neutral receptor. In the course of the determination, we observed a single-crystal phase transition, and now report on the structure of 1d, the first of a 10.10.10 cryptand,8 and this unusual occurrence of polymorphism.
Crystals of 1d (from light petroleum) were prismatic, but most of them displayed a peculiar form, with a large recess on one of the faces of the prism, often reducing the crystal to a hollow ‘box’ open on one side. A similar crystal habit has been observed for triethylenetetramine [H2N(CH2)2- NH(CH2)2NH(CH2)2NH2] and could be attributed to the presence of N[(CH2)2NH2]3, a by-product of the synthesis, which selectively inhibited the development of certain faces.9
At ambient temperature crystals of 1d are monoclinic
(α-1d), but undergo a sharp phase transition at 207 K. Of
several crystals tested, all crumbled to powder or converted to
polycrystalline blocks at this temperature, except one which was initially
of exceptionally good quality and on which a full structure determination
was performed at 210 K in space group I2/a (a
non-standard setting of C2/c, no. 15). At the moment of
transition (monitored by rotational diffractograms taken after every 1 K
cooling step) a part of the crystal split off and the remainder appeared to
become a non-merohedral twin. Its reflections were indexed on a triclinic
lattice (β-1d), which was confirmed by a full structure
determination at 150 K in space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2). No
further phase transformations were observed on cooling to 100 K.
(no. 2). No
further phase transformations were observed on cooling to 100 K.
The molecular structure of 1d in the α and β phases is shown in Fig. 1 (ESI†). In the former, monoclinic phase, the molecule lies across a crystallographic twofold axis, which is perpendicular to the N⋯N′ vector. Two of the decamethylene chains, C(1)– C(10) and C(1′)–C(10′), are related by this axis. In each chain, two atoms [C(7) and C(8) or their equivalents] are disordered over two positions with occupancies of 0.779(8) and 0.221(8). The third decamethylene chain is bisected by the twofold axis, and its four central C atoms are equally disordered over two conformations which are symmetrically related by this axis, but neither of which possesses C2 symmetry. In the β phase the molecule has no crystallographic symmetry. The average volume per non-hydrogen atom, 24 Å3 (α-1d) or 23 Å3 (β-1d), is considerably higher than the typical value for an organic compound of 18 Å3, but close to the value of 22 Å3 found in the nearest structurally studied analogue, N[(CH2)4]3 N.10,11
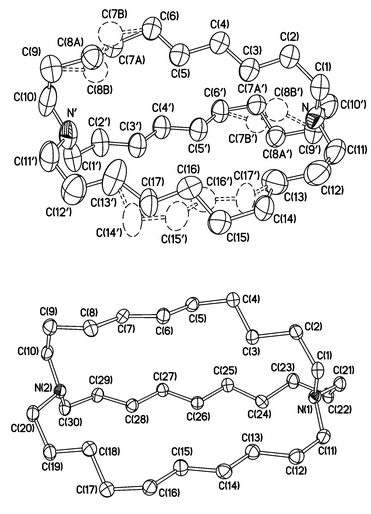 | ||
| Fig. 1 Molecular structure of 1d in the monoclinic (α, top) and triclinic (β, bottom) polymorphs, shown in the same crystallographic aspect and scale. Displacement ellipsoids are drawn at the 50% probability level. In α-1d, primed atoms are related to their equivalents by the operation of the twofold rotation axis. H atoms are omitted. | ||
In both structures the lone pairs on the nitrogen atoms point into the cavity, the pro-cryptate ‘in-in’ conformation of macro- bicyclic hosts.12 The bond distances are not unusual, while the torsion angles within the chains represent anti or gauche conformations and their patterns are different in the two structures. The molecule is slightly more elongated in the β than in the α phase, the transannular N⋯N distances being 9.14 and 9.01 Å, respectively. No significant empty volume was found in either structure or in any disorder component.13
As such topotactic phase changes between single crystal phases are relatively rare for molecular crystals, we sought to identify and describe the transformation between the high temperature α form and the low temperature β form, as illustrated in Fig. 2. The β phase is generated from the α form by first transforming the I-centered monoclinic lattice onto its primitive trinclinic one by application of the transformation matrix (−½ ½ − ½ / ½ ½ − ½ / 0 0 −1), then by an origin shift of ca. a′/2, and finally a considerable shear of the lattice, roughly along the c′ (=c) axis, widening all three lattice angles by 7–13°. In both structures the molecules are packed in layers, parallel to the (0 1 0) plane in α-1d or the (1 1 0) plane in β-1d (Fig. 2 shows the projections on these planes). The intramolecular N⋯N vector is coplanar with the layer, rigorously in α-1d, and within 3° in β-1d. The interlayer separations are essentially equal in the α and β forms (8.32 and 8.36 Å, respectively). In either case the adjacent layers are shifted against each other along the c axis (which forms the same angle of 15.5° with the N⋯N vector). In α-1d the shift is exactly c/2 (ca. 7 Å), each molecule fitting the gap in the adjacent layer. In β-1d the shift is only 4 Å (ca. 0.3c) and the molecule adopts an asymmetric conformation to accommodate to a partial overlap.
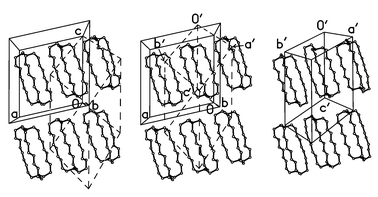 | ||
| Fig. 2 The phase transformation of 1d. Left: the structure of α-1d and the primitive triclinic cell (dashed lines, origin at 0,0,1); middle: the same with the triclinic cell (primed labels) shifted by −a′/2; right: the structure of β-1d. | ||
Although this degree of reorganization could be considered incompatible with a topotactic transition, it is not unprecedented: Gougoutas has identified examples where considerable molecular motions appear to be necessary to effect the observed phase transformations.14 The difficulties we encountered in retaining singularity suggest that in the present case we were operating at the limits of topotaxy.
The unit cell parameters of 1d change remarkably between 210 and 293 K (Table 1), while the Laue symmetry and systematic absences remain the same. With increasing temperature, the unit cell actually contracts in the direction of the twofold axis (b) by 0.286 Å (1.72%) while expanding in the perpendicular directions, again implying substantial conformational changes. Unfortunately, at room temperature the crystals decay completely in the X-ray beam within a few hours, hence a full structure determination was not possible.
| a CCDC 182/1857. See http://www.rsc.org/suppdata/cc/b0/b008044f/ for crystallographic files in .cif format. | ||||
|---|---|---|---|---|
| T/K | 293 | 210 | 150 | 120 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | I2/a | I2/a |
P |
P |
| a/Å | 14.083(6) | 13.515(1) | 10.209(1) | 10.165(6) |
| b/Å | 16.357(4) | 16.643(1) | 12.334(5) | 12.311(5) |
| c/Å | 14.398(5) | 13.934(1) | 13.904(1) | 13.876(3) |
| α/° | 90 | 90 | 63.78(1) | 63.95(2) |
| β/° | 106.93(5) | 103.12(1) | 74.73(1) | 74.69(3) |
| γ/° | 90 | 90 | 70.54(1) | 70.44(4) |
| U/Å3 | 3173(1) | 3052.4(4) | 1466.9(3) | 1456(1) |
| Z | 4 | 2 | ||
| Total reflections | 8874 | 8612 | ||
| Unique reflections | 2696 | 4916 | ||
| R int | 0.045 | 0.064 | ||
| Data with I>2σ(I) | 1547 | 3394 | ||
| R[I>2σ(I)] | 0.084 | 0.065 | ||
| wR(F2), all data | 0.254 | 0.164 | ||
Organic polymorphism is a frequently observed but poorly understood phenomenon which is rarely examined in detail.15 Despite this, it is of substantial relevance to biomolecular and pharmaceutical sciences.16 Here we have sought to describe an occurrence of polymorphism in the first structurally characterized 10.10.10 cryptand, requiring a significant degree of molecular reorganization. The historically2 and practically7 important Simmons and Park cryptand 1d has also been shown to reside in the ‘in-in’ pro-cryptate macrobicycle conformation in the solid state.
Acknowledgements
This work was supported in part by the EPSRC, and the authors are grateful to the University of Durham for provision of the X-ray and computational facilities and to Professor J. A. K. Howard for helpful discussions.Notes and references
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495 CrossRef CAS.
- H. E. Simmons and C. H. Park, J. Am. Chem. Soc., 1968, 90, 2428 CrossRef CAS; C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2429 CrossRef CAS; C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431 CrossRef CAS.
- Comprehensive Supramolecular Chemistry, ed. G. W. Gokel, Elsevier, Oxford, 1996, vol. 1. Search PubMed.
- Cambridge Structural Database Version 5.17 (197481 structures, April 1999 release): F. H. Allen and O. Kennard, Chem. Des. Autom. News, 1993, 8, 1 Search PubMed; F. H. Allen and O. Kennard, Chem. Des. Autom. News, 1993, 8, 31 Search PubMed.
- R. A. Bell, G. G. Christoph, F. R. Fronczek and R. E. Marsh, Science, 1975, 190, 151 CAS.
- Cf. melting points, 1a, 35 °C; 1b, 120 °C; 1c, 32 °C; 1d, 114 °C (from ref. 2). See: J. Dale, Angew. Chem., 1966, 5, 1000 for a discussion of the effects of chain length on conformational stability in cyclic structures..
- This cryptand has been used in a number of anion complexation and transport studies, see: B. Dietrich, T. M. Fyles, M. W. Hosseini, J.-M. Lehn and K. C. Kaye, J. Chem. Soc., Chem. Commun., 1988, 691 RSC; J.-P. Kintzinger, J.-M. Lehn, E. Kauffmann, J. Dye and A. I. Popov, J. Am. Chem. Soc., 1983, 105, 7549 CrossRef CAS; E. Graf and J.-M. Lehn, J. Am. Chem. Soc., 1976, 98, 6403 CrossRef CAS.
- The crystal structure of a 10.10.10 macrobicycle with tin at the bridgeheads has been published. However, this can not be considered a cryptand due to the tetravalent nature nature of tin and the consequent, forced out–out conformation. See: J. H. Horner, P. J. Squatritto, N. McGuire, J. P. Riebenspies and M. Newcomb, Organometallics, 1991, 10, 1741 CrossRef CAS.
- I. Bernal, personal communication..
- R. W. Alder, A. G. Orpen and R. B. Sessions, J. Chem. Soc., Chem. Commun., 1983, 999 RSC.
- W. P. Schaefer and R. F. Marsh, J. Chem. Soc., Chem. Commun., 1984, 1555 RSC.
- R. W. Alder and S. P. East, Chem. Rev., 1996, 96, 2097 CrossRef CAS.
- A. L. Spek, PLATON, a multi-purpose crystallographic tool. University of Utrecht, The Netherlands..
- J. Z. Gougoutas, Pure Appl. Chem., 1971, 27, 305 CAS.
- C. N. R. Rao and K. J. Rao, Phase Transitions in Solids, McGraw-Hill, London, 1978; Search PubMed; A. Gavezzotti and M. Simonetta, Chem. Rev., 1982, 82, 1 Search PubMed; J. D. Dunitz, Acta Crystallogr., Sect. B, 1995, 51, 619 CrossRef CAS; M. R. Caira, Top. Curr. Chem., 1998, 198, 163 CrossRef.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental details of the crystal structure determinations, orientation and phase transformation matrices and diagram of the unit cells of α- and β-1d. See http://www.rsc.org/suppdata/cc/b0/b008044f/ |
| ‡ Present address: Department of Chemistry and Biochemistry, University of California, Los Angeles, 405 Hilgard Ave., Los Angeles, CA 90095, USA. E-mail: mascal@chem.ucla.edu. |
| This journal is © The Royal Society of Chemistry 2001 |