Aluminium-containing ring systems and N-heterocycle formation via nitrile insertions into Al–N bonds
Vernon C. Gibson*, Carl Redshaw†, Andrew J. P. White and David J. Williams
Department of Chemistry, Imperial College, South Kensington, London, UK SW7 2AY. E-mail: v.gibson@ic.ac.uk
First published on 15th December 2000
Abstract
Reactions of Me3Al with 1,2-diaminobenzene [1,2- (H2N)2C6H4] or anthranilic acid, [1,2-(H2N)(HO2- C)C6H4], followed by treatment with acetonitrile, afford tetranuclear and hexanuclear aluminium-containing ring systems; a single crystal X-ray structure on the hexametallic product reveals the construction of quinazoline ligands arising via insertion of acetonitrile into Al–N bonds.
It is well over one hundred years since the trimerisation of nitriles to triazines by organometallic reagents was first noted, by Hofmann1 using sodium, and by Frankland2 in his studies on Et2Zn. During the 1960s, Wade3 and Lappert4 did much to advance the understanding of nitrile binding and insertion reactions at main group centres; however, the potential of such reactions for synthesising useful heterocycles and interesting metal-containing ring systems has remained largely undeveloped.
Recently, we described the synthesis of large aluminium-containing ring systems via treatment of Me3Al with hydrazines;5 these included a highly unusual octa-aluminium structural analogue of a tetrapyrrole. The key macrocycle-forming step revolves around the insertion of acetonitrile into the aluminium–nitrogen bonds of intermediate amide species. With a view to probing the generality of this approach for the synthesis of aluminium-containing macrocycles, and also to evaluate the potential of this methodology for constructing nitrogen heterocycles, we have extended the study to other classes of amine substrate. Here, we describe the reactivity of Me3Al towards 1,2-diaminobenzene [1,2-(H2N)2C6H4] and anthranilic acid [1,2-(H2N)(HO2C)C6H4]. The former gives rise to a tetranuclear complex, the latter to an unprecedented hexanuclear species incorporating N-heterocyclic ligands.
Slow addition of a solution of Me3Al (2 equivalents) in toluene to 1,2-(H2N)2C6H4, followed by a 12 h reflux, afforded a pale brown solution. After removal of the solvent under reduced pressure, the residue was dissolved in acetonitrile and heated to reflux for 2–3 min. Slow cooling of this solution, followed by standing at room temperature for 2 days, gave colourless needles of 1 in 40% yield (Scheme 1). The 1H NMR spectrum‡ of 1 consists of four equal intensity singlets in the Al–methyl region (δ −0.37 to −0.75) together with a similar intensity singlet at δ 1.29 attributable to carbon-bonded methyl groups. X-Ray analysis§ reveals the product to be the Ci symmetric twelve-membered macrocyclic complex 1 comprising four aluminium atoms (two bridging and two chelating), six nitrogen atoms and two carbon atoms, The structure of this product is closely related to that obtained from the reaction of MePhNNH2 with Me3Al,5 and is not described in further detail here.
 | ||
| Scheme 1 Compounds 1–4 (inserted molecules of acetonitrile are shown in bold). | ||
In the absence of acetonitrile, the 2∶1 reaction of Me3Al and 1,2-(H2N)2C6H4 (in toluene) has been shown to afford the asymmetric complex [(Me2Al)2AlMe(C6H4(NH)2 )2]·AlMe32.6 This, or a closely related derivative, is the likely precursor to 1. Formation of the 12-membered ring product is brought about by insertion of two acetonitrile molecules into the Al–N bonds, in a related manner to that postulated for the reaction of hydrazines with Me3Al/MeCN.5
Similar treatment of anthranilic acid, [1,2-(NH2)- (HO2C)C6H4] (twice sublimed) with 2 equivalents of a toluene solution of Me3Al affords, after work-up in acetonitrile, large yellow prisms for which the 1H NMR spectrum possesses nine distinct singlets in the Al–methyl region. The X-ray analysis§ of the product revealed the chiral trimeric hexanuclear complex [(Me2AlL)(MeAl)(μ3-O)(μ-O)]}33 (L = quinazoline) shown in Fig. 1. The central Al3O3 ring has a twisted boat conformation, whereas the three attached Al2O2CN rings each have a folded envelope geometry. All six aluminium atoms exhibit marked departures from ideal tetrahedral geometry with angles in the range 100.9(2)–124.4(3)°. Although not possessing strict C3 symmetry [the methyls on Al(2) and Al(4) are ‘up’, whilst that on Al(6) is ‘down’], the pattern of bonding throughout the structure is essentially three-fold symmetric. It is interesting to note that whilst all of the Al–O bond lengths within the central Al3O3 ring and also those to O(2), O(4) and O(6) are all essentially the same [1.770(4)–1.794(4) Å], those to Al(1), Al(3) and Al(5) are all significantly longer [1.811(4)–1.827(4) Å]. As 3 is not the product of a chiral synthesis, the presence in the crystals of molecules of only one chirality is a consequence of spontaneous resolution upon crystallisation.
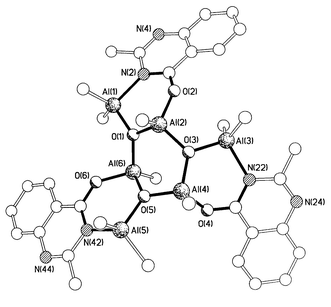 | ||
| Fig. 1 The molecular structure of 3. Selected bond lengths (Å); Al(1)–O(1) 1.811(4), Al(1)–N(2) 2.021(5), Al(2)–O(1) 1.785(4), Al(2)–O(3) 1.788(4), Al(2)–O(2) 1.779(4), Al(3)–O(3) 1.827(4), Al(3)–N(22) 2.034(5), Al(4)–O(3) 1.770(4), Al(4)–O(4) 1.783(5), Al(4)–O(5) 1.785(4), Al(5)–O(5) 1.825(4), Al(5)–N(42) 2.015, Al(6)–O(1) 1.784(4), Al(6)–O(5) 1.794(4), Al(6)–O(6) 1.785(5). | ||
Whereas the formation of 1 can be rationalised in terms of acetonitrile insertion into the aluminium–nitrogen bonds of 2, the carboxylic acid group of anthranilic acid contributes oxygen atoms to the Al3O3 core around which the quinazoline ligands are clustered. The reaction reproducibly affords 3 in ca. 45% yield; its hydrolysis then readily releases the 2-methyl-4(3H)-quinazolinone heterocycle 4 (identified by comparison of NMR data with those of an authentic sample) in excellent yield. We note that the formation of N-heterocycles by this methodology is related to the intramolecular Ritter reaction7–9 in which nitrilium salts, generated in the presence of Friedel–Crafts reagents, react with a second nitrile molecule to give quinazoline ring systems.9
Future studies will focus on further exploiting nitrile insertions into aluminium (and gallium) bonds to access unusual inorganic ring systems and nitrogen heterocycle products.
Acknowledgements
The EPSRC and the Leverhulme Trust (for a Research Fellowship to C. R.) are thanked for financial support. Professor Charles Rees is thanked for helpful discussions.Notes and References
- A. W. Hofmann, Chem. Ber., 1868, 1, 194 Search PubMed.
- E. Frankland and J. C. Evans, J. Chem. Soc., 1880, 37, 563 Search PubMed.
- H. J. Emeléus and K. Wade, J. Chem. Soc., 1960, 2614 RSC; J. E. Lloyd and K. Wade, J. Chem. Soc., 1964, 1649 RSC; J. E. Lloyd and K. Wade, J. Chem. Soc., 1965, 2662 RSC; J. R. Jennings, J. E. Lloyd and K. Wade, J. Chem. Soc., 1965, 5083 RSC; I. Pattinson and K. Wade, J. Chem. Soc., 1968, 57 Search PubMed; J. R. Jennings and K. Wade, J. Chem. Soc. A, 1968, 1946 RSC.
- W. Gerrard, M. F. Lappert and J. W. Wallis, J. Chem. Soc., 1960, 2178 RSC; M. F. Lappert and B. Prokai, Adv. Organomet. Chem., 1967, 5, 225 and references therein. Search PubMed.
- V. C. Gibson, C. Redshaw, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 961 CrossRef CAS.
- R. L. Wells, H. Rahbarnoohi, P. B. Glaser, L. M. Liable-Sands and A. L. Rheingold, Organometallics, 1996, 15, 3204 CrossRef CAS.
- J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045 CrossRef CAS; J. J. Ritter and J. Kalish, J. Am. Chem. Soc., 1948, 70, 4048 CrossRef.
- L. I. Krimen and D. J. Cota, Org. React. (N.Y.), 1969, 17, 213 Search PubMed.
- R. Bishop, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon,Oxford, UK, 1991, vol. 6 and references therein. Search PubMed.
Footnotes |
| † Present address: School of Chemical Sciences, University of East Anglia, Norwich, UK NR4 7TJ. |
| ‡ Satisfactory microanalyses
have been obtained. For 1: 1H NMR (400 MHz, C6D6, 298 K), δ −0.75, −0.48, −0.42, −0.37 (4 × s, 8 × 3H, AlMe), 1.29 (s, 2 × 3H, CMe), 6.36–7.27 (3 × m, 8H, aryl H), NH not seen. 13C NMR (100.6 MHz, C6D6, 298 K), δ −12.24, −12.81 (br, AlMe), −8.91 (br, AlMe), −5.99 (br, AlMe). IR: ν(N–H) 3247 cm−1. For 3: 1H NMR (400 MHz, CDCl3, 298 K), δ −0.87, −0.82, −0.59, −0.47, −0.46, −0.45, −0.24, −0.23, −0.22 (9 × s, each 3H, AlMe), 2.79 (s, 3H, Me-quin), 2.88 (s, 3H, Me-quin), 2.89 (s, 3H, Me-quin), 7.61 (m, 3H, quinH), 7.82 (m, 3H, quinH), 7.92 (m, 3H, quinH), 8.35 (m, 3H, quinH). 13C NMR (100.6 MHz, CDCl3, 298 K), δ −12.24, −11.56 (2 × m, 3 × AlMe), −7.22, −6.79, −6.45, −5.43, −5.10, −4.57 (6 × s, 3 × AlMe2), 25.53, 25.82, 25.93 (3 × s, Me-quin), 117.15 (m, 3 × aryl C), 125.28–127.61 (overlapping m, 9 × aryl C), 136.33 (m, 3 × aryl C), 150.98 (m, 3 × aryl C), 158.45 (m, 3 × aryl C), 169.31 (m, 3 × aryl C). EI-MS: m/z: 807 (M+ − Me). IR: ν(μ3-O)Al3 800 cm−1. |
| § Crystal data: For
1: C24H42N6Al4,
M = 522.6, orthorhombic, space group Pbca (no. 61),
a = 8.717(1), b = 16.868(1), c = 20.416(2)
Å, V = 3002.0(4) Å3, Z = 4 (the
complex has crystallographic Ci symmetry),
Dc = 1.156 g cm−3,
μ(Cu-Kα) = 16.1 cm−1, F(000) =
1120, T = 183 K; clear prisms, 0.27 × 0.23 × 0.13 mm,
Siemens P4/RA diffractometer, ω-scans, 2182 independent
reflections. The structure was solved by direct methods and the
non-hydrogen atoms were refined anisotropically using full-matrix least
squares based on F2 to give
R1 = 0.069, wR2 =
0.173 for 1479 independent observed reflections
[|Fo| >
4σ(|Fo|),
2θ < 120°] and 162 parameters. For 3: C36H48N6O6Al6, M = 822.7, orthorhombic, space group P212121 (no. 19), a = 12.152(1), b = 16.054(1), c = 22.310(1) Å, V = 4352.3(6) Å3, Z = 4, Dc = 1.256 g cm−3, μ(Cu-Kα) = 17.9 cm−1, F(000) = 1728, T = 173 K; yellow rhombs, 0.50 × 0.23 × 0.23 mm, Siemens P4/RA diffractometer, ω-scans, 4017 independent reflections. The structure was solved by direct methods and the non-hydrogen atoms were refined anisotropically using full-matrix least squares based on F2 to give R1 = 0.055, wR2 = 0.133 for 3483 independent observed reflections [|Fo| > 4σ(|Fo|), 2θ < 128°] and 488 parameters. The absolute structure of 3 was determined by use of the Flack parameter which refined to a value of −0.07(7). CCDC 182/1855. See http://www.rsc.org/suppdata/cc/b0/b007810g/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |