Metal ion catalysis in chemisorption and dehydrogenation of alkanes on aluminium hydroxide clusters, revealed by theoretical calculations
Dan
Fǎrcasiu
* and
Povilas
Lukinskas
Department of Chemical and Petroleum Engineering, University of Pittsburgh, 1249 Benedum Hall, Pittsburgh, PA 15261, USA.. E-mail: dfarca@pitt.edu
First published on 14th December 2000
Abstract
Electron-correlated DFT calculations with a large basis set show that propane adds to coordinatively unsaturated aluminium, as in the clusters (HO)3Al(OH2)x (x = 0, 1), by aluminium insertion into a C–H bond, followed by hydrogen migration to an oxygen atom and predict correctly experimental observations; the alternative pathway involving alkyl–oxygen interaction has a much higher energy barrier and does not predict correctly the experimental results.
Following the progress of superacid chemistry, the reactions of hydrocarbons on aluminosilicates, including zeolites, have been interpreted as involving the activation by hydron transfer to form carbocations as intermediates or transition structures.1 This type of activation, characteristic of superacids in solutions, e.g. HF–SbF5,2 HF–TaF5,3 or H2O–AlX3,4 is not found for trifluoromethanesulfonic acid (TFMSA), which activates alkanes by an oxidative mechanism.5 Yet, TFMSA is a much stronger acid than the zeolite catalysts.6 This paradox, observable to an outsider, has been lost to the researchers in the field.
The computations on the activation of alkane C–H bonds have attempted to describe the accepted mechanism and sought mostly pathways based on hydron transfer.7 The same held for the hydrogen chemisorption on alumina, responsible for the high-temperature H2/D2 exchange,8 which was thought to involve heterolysis of the H–H bond, with the hydron going to the oxygen (base) and the hydride to aluminium (acid).9
Standard ab initio (MP2) and DFT (B3LYP) geometry optimizations with large basis sets and search of the reaction coordinate without the imposition of a pathway have shown, however, that hydrogen chemisorption occurs through the interaction of H2 with the aluminium (metal ion catalysis) until both hydrogen atoms are bonded to Al, after which one hydrogen migrates to oxygen.10 The reactivity of aluminium centers varied in the order: tri- > tetra- > penta-coordinated and the tetracoordinated aluminium in a silica-alumina cluster chemisorbed hydrogen by the same mechanism.10b
Alumina catalyzes the H–D exchange of saturated hydrocarbons as well,11 a reaction also classified as acid–base catalysis.11 Like the hydrogen chemisorption, the reaction of methane with Al(OH)3 was described as a heterolytic reaction with an acid–base pair on the surface, with a hydron going to the negative oxygen and a methyl anion to the metal.12 A computational search in which the reaction pathway was not presupposed was, therefore, in order. We studied the reaction of propane with aluminium hydroxide clusters, (HO)3Al(OH2)x (1, x = 0; 2, x = 1).
B3LYP/6-31G** geometry optimizations and frequency analyses (giving also the zero point energy corrections, ZPE),13 transition structure searching by the STQN (Synchronous Transit-Guided Quasi Newton) method,14 and reaction pathway identification by intrinsic reaction coordinate (IRC)15 tracking were conducted with the Gaussian 98 program,16 as described before.10 Pathway b (defined below) for chemisorption on 1 was also examined by MP2/6-31G**, to check the agreement between the two methods (as done in previous work).10a To model the alumina surface, the geometry of the tricoordinated aluminium reactant 1 was the optimized geometry of 2, with the water ligand removed.10 In the reaction of 1 with propane, the O1–O2–Al–O3 dihedral angle was kept constant. The alternative of freezing the outer atoms of the cluster and allowing the aluminium atom a breathing movement gave a similar potential energy barrier (PEB) for H2 chemisorption.10a Our goal was to explore the existence of a reaction pathway, rather than to determine accurately the relative energies of reactants, intermediates, and transition structures.
Weak complexes of propane with 1 (3) and 2 (4) were located and two types of chemisorption products were identified, with C–Al bond (5 and 6, respectively) and with C–O bond (7 and 8, respectively), shown in eqns. (1) and (2). The relative reactivities of primary and secondary C–H bonds were also tested [series a and b in eqns. (1) and (2)]. The O–alkyl complexes were less stable than the Al–alkyl complexes by 5–7 kcal mol−1.

| (1) |
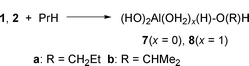
| (2) |
All chemisorption products react further to form hydrogen and propene, complexed with the aluminium cluster. The energies of intermediates and products, relative to the starting materials, are shown in Table 1.
| Catalyst cluster | Reaction pathway | Physisorbed reactant | TS1 | Chemisorbed complex | TS2 | Physisorbed product(s) | Isolated products |
|---|---|---|---|---|---|---|---|
| a B3LYP/6-31G**//B3LYP/6-31G**, kcal mol−1, relative to the isolated starting materials (1 or 2 and PrnH). b 1 cal = 4.184 J. c 1 → 5a, eqn. (1). d 1 → 5b, eqn. (1). e MP2(FC)/6-31G**// MP2(FC)/6-31G** values are −6.33, 33.59 and 31.50 kcal mol−1, respectively. f 1 → 7b, eqn. (2). g Propene chemisorbed on the (H2O)2AlH–OH2 cluster. h 46.65 kcal mol−1 if (H2O)2AlH–OH2 is a product. i 2 → 6a, eqn. (1). j d(Al–OH2) = 2.11 Å, see text. k d(Al–OH2) = 2.31 Å, see text. l 2 → 6b, eqn. (1). m Decomposition to 5b occurred. n 2 → 8b, eqn. (2). o d(Al–OH2) = 2.00 Å, see text. p d(Al–OH2) = 2.20 Å, see text. q 48.14 kcal mol−1 if the hydrogenated cluster is a product. | |||||||
| 1 | Al–CH2Etc | −4.01 | 32.19 | 31.24 | 57.10 | 17.63 | 29.23 |
| 1 | Al–CHMe2d | −4.01e | 35.14e | 33.54e | 62.41 | 17.63 | 29.23 |
| 1 | O–CHMe2f | −4.01 | 72.31 | 37.34 | 70.35 | 34.77g | 29.23h |
| 2 | Al–CH2Eti | −1.93 | 43.95 | 25.59 | 74.20,j 72.21k | 26.56 | 29.23 |
| 2 | Al–CHMe2l | −1.93 | —m | — | — | — | — |
| 2 | O–CHMe2n | −1.93 | 82.22 | 32.28 | 75.98,o 67.31p | 43.77 | 29.23q |
It is seen that chemisorption and dehydrogenation are catalyzed by both tricoordinated and tetracoordinated aluminium centers. The former are more reactive, just as for the hydrogen chemisorption.10 For the aluminium–alkyl pathway, eqn. (1), a primary C–H bond (a) is more reactive than a secondary C–H bond (b). Only option b was studied for the O–alkyl pathway [eqn. (2)], because the alkyl group acquired a positive charge. On the aluminium–alkyl pathway, the barrier for alkane chemisorption (TS1) is much smaller than the barrier (TS2) for the dehydrogenation step, whereas in the oxygen–alkyl pathway, TS1 is higher in energy than TS2. The O–alkyl pathway is higher in energy than the aluminium–alkyl pathway. In the second step of the reaction on 2, the cleavage of an Al–O bond is easier than the elimination of propene. If the cluster was part of a solid, the lattice rigidity would determine the degree of Al–O bond separation. Therefore, the elimination step was followed at two lengths of the labile Al–O bond: the same as in the intermediate (6 or 8) and extended by 0.2 Å. Both values are given in Table 1, for each reaction pathway; the latter are smaller.
The transition structure for the chemisorption step on the aluminum–alkyl pathway,14 shown in Fig. 1 (left)17 for the reaction 1 → 5a, was similar to that for hydrogen chemisorption. The imaginary frequency was the bending of the Al–H bond toward O (migration of hydrogen from Al to O).18 Thus, the reaction mechanism consists of the insertion of aluminium into the C–H bond, followed by hydrogen migration from Al to O, just as for hydrogen chemisorption.10 In the elimination step, there is less cleavage of the Al–C bond than of the C–H bond at the transition state (Fig. 1, right).19 For the elimination step of the O–alkyl pathway, the hydrogen is transferred to another oxygen atom.
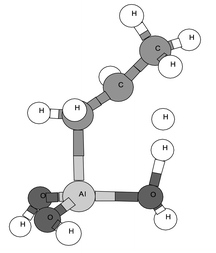 | ||
| Fig. 1 Left: transition structure (TS1) for the chemisorption of propane (primary C–H) on a tricoordinated aluminium cluster ( 1 → 5a). Right: transition structure (TS2) for the elimination of propane from the chemisorbed complex 5a. | ||
The reactivity order prim > sec and the prediction that hydrogen exchange [reverse of eqn. (1)] is faster than further reaction of the olefin (e.g. on acid sites) agrees with the experiment. (The exchange of H2 with the clusters has lower barriers than the exchange of the C–H bonds.10) The described mechanism is relevant for the hydrogen exchange11,19 and alkene hydrogenation/alkane dehydrogenation.20 These reactions have been described as catalyzed by Brønsted acid centers, with carbocations as intermediates or transition structures.7,9,12,21 The cleavage of H–H and C–H bonds by insertion of metal atoms and ions (metal and metal ion catalysis), known for heavy metals, particularly noble metals, was not considered for aluminium. We show now that Al(O–)n sites with n = 3 and 4, are active in metal ion catalysis. An increase in reactivity for ‘broken lattices’ of zeolites or for extraframework aluminium species in steamed zeolites is predicted.
Acknowledgements
This research was supported by the grant CTS-812704, from NSF, and by a grant of supercomputer time from NCSA, in Urbana, IL.Notes and references
- See the discussion in W. K. Hall, E. A. Lombardo and J. Engelhardt, J. Catal., 1989, 115, 611 CrossRef CAS.
- D. M. Brouwer and H. Hogeveen, Prog. Phys. Org. Chem., 1972, 9, 179 Search PubMed.
- D. Fǎrcasiu, M. Siskin and R. P. Rhodes, J. Am. Chem. Soc., 1979, 101, 7671 CrossRef CAS.
- C. D. Nenitzescu and I. P. Cantuniari, Ber. Dtsch. Chem. Ges., 1933, 66, 1097 Search PubMed; H. S. Bloch, H. Pines and L. Schmerling, J. Am. Chem. Soc., 1946, 68, 153 CrossRef CAS.
- D. Fǎrcasiu and P. Lukinskas, J. Chem. Soc., Perkin Trans. 2, 1999, 2715 RSC; D. Fǎrcasiu and P. Lukinskas, J. Chem. Soc., Perkin Trans. 2, 2000, 2295 RSC.
- T. Xu, E. J. Munson and J. F. Haw, J. Am. Chem. Soc., 1994, 116, 1962 CrossRef CAS; D. Fǎrcasiu, J. Chem. Soc., Chem. Commun., 1994, 1801 RSC.
- S. P. Bates and R. A. van Santen, Adv. Catal., 1998, 42, 1 Search PubMed; P. M. Esteves, M. A. C. Nascimento and C. J. A. Mota, J. Phys. Chem., B, 1999, 103, 10417 CrossRef CAS.
- H. S. Taylor and H. Diamond, J. Am. Chem. Soc., 1935, 57, 1256 CrossRef CAS.
- I. N. Senchenya and V. B. Kazanskii, Kinet. Katal., 1988, 29, 1331 Search PubMed; F. Ioka, T. Sakka, Y. Ogata and M. Iwasaki, Can. J. Chem., 1993, 71, 663 CAS.
- (a) D. Fǎrcasiu and P. Lukinskas, J. Phys. Chem., A, 1999, 103, 8483 CrossRef CAS; (b) P. Lukinskas and D. Fǎrcasiu, Appl. Catal. A, in press. Search PubMed.
- For references, see: S. Yoshida, in Theoretical Aspects of Heterogeneous Catalysis, ed. J. B. Moffat, Van Nostrand Reinhold, New York, 1990, p. 506. Search PubMed.
- M. J. Capitán, J. A. Odriozola, A. Márquez and J. F. Sanz, J. Catal., 1995, 156, 273 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley-Interscience, New York, NY, 1986. Search PubMed.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449 Search PubMed.
- C. Gonzales and H. B. Schlegel, J. Phys. Chem., 1989, 90, 2154 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Daprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, P. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.1, Gaussian, Inc., Pittsburgh PA, 1998. Search PubMed.
- G. Schaftenaar, MOLDEN. A Portable Electron Density Program, available at: ftp ftp.caos.kun.nl Name: anonymous..
- Projections generated with: Xmol, version 1.3.1, Minnesota Supercomputing Center, Inc., Minneapolis, MN, 1993..
- B. Schoofs, J. Schuermans and R. A. Schoonheydt, Microporous Mesoporous Mater., 2000, 35–36, 99 CrossRef CAS.
- V. C. F. Holm and R. W. Blue, Ind. Eng. Chem., 1951, 43, 501 Search PubMed.
- S. Senger and L. Radom, J. Am. Chem. Soc., 2000, 122, 2613 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |