DOI:
10.1039/B007685F
(Paper)
Analyst, 2001,
126, 109-113
Determination of Co(II) by chemiluminescence
after in situ electrochemical pre-separation on a flow-through
mercury film electrode
Received 21st September 2000, Accepted 26th October 2000
First published on 19th December 2000
Abstract
A novel method of electrochemical pre-separation of
Co(II) before detection by chemiluminescence is reported
together with the associated instrumentation. The Co(II) ions
were selectively pre-separated on a mercury film electrode (MFE) by on-line
reduction, then the accumulated metal was oxidised and selectively stripped
back into the flowing solution as Co(II). These secondary ions
were quantified as a result of their catalytic activity on the
chemiluminescent reaction between luminol and hydrogen peroxide that was
also induced on-line. The whole sequence was carried out in an automated
flow-through system, in which the electrochemical pre-separation of metals
was performed in either continuous flow or flow injection analysis (FIA)
regimes. The scope of the method, both in terms of selectivity and
sensitivity, has been demonstrated and the quantitative determination of
Co(II) by the proposed method has been investigated. For a
period of continuous flow pre-separation of 4 min, the calibration curve
for Co(II) was linear up to a concentration of 100 μg
l−1, the relative standard deviation was 4% at the 20
μg l−1 level and the limit of detection was 0.5 μg
l−1 (at the 3σ level). The method was
applied to the determination of the cobalt content in a high purity iron
sample.
Introduction
Chemiluminescence (CL) is an extremely sensitive analytical technique
used for the determination of both organic and inorganic species. The high
sensitivity of CL makes it a particularly attractive choice for the
quantification of trace metals in various samples.1–3 The utility of the method is based on the
catalytic (or inhibitory) effect of many metal ions on a suitable CL
reaction. The most widely exploited, but by no means the only, such
reaction is the oxidation of luminol by hydrogen peroxide in an alkaline
solution. Within a certain concentration range, the catalytic CL intensity
is proportional to the concentration of the metal ion in the sample.
However, CL is not a selective method since several metal ions catalyse (or
inhibit) most chemiluminescent reactions to varying degrees. As a
consequence of this drawback, an on-line separation step is required prior
to the detection stage. Traditionally, various methods have been employed
to achieve this separation including selective complexation,4 liquid–liquid extraction, membrane
separations and, by far the commonest approach, ion exchange
columns.3 Recently, a method has been
reported that made use of an electrochemical pre-separation stage on a gold
electrode for the determination of traces of Cu(II) by
chemiluminescence.5,6In this paper, we report a novel method that makes use of
electrochemical pre-separation on a flow-through electrochemical (EC) cell
equipped with a MFE in order to achieve the required selectivity and
sensitivity for trace analysis. The MFE combines the relative advantages of
the solid and mercury drop electrodes and is ideal for flow-through
applications.7 The principle of the method
was that the target metal ion was pre-separated on-line by reduction on the
MFE. Following this separation stage, the metal was oxidised and stripped
back again into the flowing carrier as a cation to be subsequently detected
by CL in an optical cell. The required instrumentation is described and the
instrumental and chemical parameters to be considered are discussed. In
particular, the selectivity was maximised by optimisation of the
pre-separation and stripping potentials while the sensitivity could be
altered by varying the pre-separation mode and the accumulation time. The
method was accessed and validated for the detection of trace
Co(II) in a reference material.
Experimental
Chemicals
All the chemicals were of analytical grade and ultrapure water
(conductivity > 18 MΩ−1 cm−1)
was used for the preparation of the solutions. A carrier carbonate buffer
solution pH 10.2 was prepared daily by mixing the appropriate amounts of
NaHCO3 and NaOH. A stock solution of luminol (10 mmol
l−1 ) was prepared every week and kept in the dark; a 1
mmol l−1 solution was prepared daily and kept in a dark
bottle during the experiments. A 10 mmol l−1 hydrogen
peroxide solution was also prepared daily from 30% w/v hydrogen peroxide.
The mercury plating solution was 1 mmol l−1
Hg(II) in 0.1 mol l−1
KNO3–0.01 mol l−1 HNO3.
Standard Co(II) standard solutions were prepared daily from a
1000 mg l−1 atomic absorption standard solution by serial
dilution. A solution of 1000 mg l−1 in
Cu(II) was prepared from CuSO4 and more dilute
solutions were prepared by serial dilution.Equipment
The automated flow system is a modified version of the automated flow
system described previously,7 in this case
optimised for CL. Both FIA and continuous flow pre-separation could be
carried out depending on the positioning of the EC flow cell. The
experimental configuration is illustrated in Fig.
1. The valves, configured either for mixing, M, or switching, I,
were micro-solenoid devices purchased from the Lee Company (Westbrook, CT,
USA). The following parameters were software adjustable: flow rates of the
solutions, number of sample injections, injection time, and delay time. For
the FIA operation, the volume of the sample loop was 100 μl. The
electrochemical cell (ECC in Fig. 1) was a thin layer cell, designed
in-house with a flow channel thickness of 0.2 mm. The working electrode was
a glassy carbon rod (3 mm in diameter) (Ringsdorf Carbon, Germany), the
reference electrode was a home-made Ag/AgCl electrode positioned opposite
the working electrode and the counter electrode was a glassy carbon rod
positioned downstream, near the outlet of the cell. The home-made optical
flow cell, positioned downstream of the EC cell, consisted of a rectangular
channel of zig-zag configuration, 10 cm in length, machined in a block of
Perspex. The potentiostat was a PARC 273 (EG&G, NJ, USA) controlled by
a 386 PC through the PARC Model 270 Electrochemical Software. The optical
detector was a Hamamatsu HC120 integrated photomultiplier
(PMT)-amplifier-power supply module operating at a voltage of 250 V. The
optical flow cell was positioned in front of the PMT window in a
light-tight box constructed in house. The output of the detector module
(i.e. the CL response) was fed simultaneously to a BBC Coertz
Metrawatt SE 120 chart recorder and to a LABPC+ data acquisition (DAQ) card
(National Instruments, Austin, TX, USA) that was interfaced to a 486 PC and
was controlled by a data acquisition program written in LabVIEW 4.0
(National Instruments). The response could be viewed on the screen of the
computer and also saved on disk. A PARC Model 303 HMDE has been used for
the voltammetric experiments with Co(II).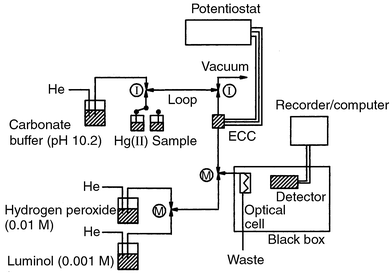 |
| | Fig. 1 Configuration of the flow system for the detection of Co(II)
by CL after EC pre-separation in the FIA mode. M and I indicate mixing
valves and injector valves, respectively. ECC represents the
electrochemical thin layer cell. In the continuous flow mode, the loop is
replaced by the ECC. | |
Experimental procedure
0.1 g of the iron sample was dissolved in 2 ml of 4 mol
l−1 M HNO3, 0.2 g of potassium tartrate was
added (in order to complex Fe(III)), the pH was adjusted to 10.2
with NaOH and the solution was diluted to 100 ml with water.The glassy carbon surface was pre-treated as described
previously.8 Then, the mercury film was
plated at −1.0 V by passing the mercury solution through the cell at
0.6 ml min−1 (continuous flow mode) for 60 s or by making
5 discrete injections of the mercury solution (FIA mode).
The potential was then switched to the pre-separation potential (usually
−1.4 V) and the pre-separation of Co(II) was initiated by
passing the sample through the cell under continuous flow at 0.6 ml
min−1 or by injecting the sample in the carrier (FIA
mode). In the FIA configuration, the sampling time required for the sample
to wash and fill the injection loop was 15 s. The accumulated cobalt was
stripped off in the flowing carbonate buffer carrier as Co(II)
by stepping to a more anodic stripping potential (ranging from −0.8
to −1.2 V), mixed with the combined luminol–hydrogen peroxide
solution and detected by CL. Finally, the MFE was regenerated
electrochemically in the carbonate buffer at a potential ranging from +0.2
to −0.8 V. The flow rates of the carrier solution and of the
luminol–hydrogen peroxide mixture were 1.5 and 1.3 ml
min−1, respectively.
Results and discussion
Study of the electrochemical pre-separation and stripping
of Co(II)
In principle, a combination of EC preconcentation and stripping prior to
the actual detection process can enhance both the sensitivity and the
selectivity of the analysis of metals. The sensitivity is improved as a
result of the enrichment of the electrode surface with the metal under
investigation. The metal is first reduced and deposited on the electrode
surface under cathodic potentiostatic conditions (i.e.
electrolysis) for a defined time period and, then, the accumulated metal is
stripped off the electrode and detected. Stripping can be accomplished
via either voltammetric techniques (involving a scan of the
working electrode potential with respect to time in the anodic direction)
or potential-step techniques (based on a step change of the potential of
the working electrode to an anodic value). The former method of stripping
is commonly exploited in anodic stripping voltammetry (ASV)9 in which the potential scan also serves as the
detection scheme. Although this method offers adequate selectivity and
sensitivity for some metals, it is hampered by drawbacks (such as
capacitive currents, the need for deoxygenation, insensitivity to
irreversible redox reactions etc.). On the other hand, with the
potential-step techniques, the selectivity towards a particular metal can
be improved by judiciously selecting the pre-separation and stripping
potentials, as is diagramatically illustrated in Fig.
2. The anodic limit, Eal, and cathodic limit,
Ecl, correspond to the potentials at which
oxidation and reduction, respectively, of the supporting electrolyte occur
and define the useful potential range within which EC pre-separation is
feasible. As long as a pre-separation potential, Ep, is
applied to the working electrode, metal ions with redox potential
E
![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) Ep will be reduced and accumulated on
the electrode while metal ions with redox potential E
⩽
Ep will be unaffected. As soon as the potential is
stepped to a more positive stripping potential, Es, the
accumulated metals with redox potentials, Ei, where
Es > Ei >
Ep will be oxidised and stripped back into the solution
while those metals with redox potentials Ei >
Es will remain on the electrode. Thus, from the
potentially useful range (Eal, Ecl)
only the metals in the narrow range (Es,
Ep) will be available for detection. In this mode of
stripping, amperometric, spectroscopic or chemiluminescent detection can be
employed. For the present work, CL detection was selected due to its
simplicity and sensitivity. Dynamic EC techniques were initially used to
study the pre-separation behaviour of Co(II) on a mercury
electrode. The EC accumulation and stripping of Co(II) was
investigated on a hanging mercury drop electrode (HMDE). ASV was initially
investigated but this technique did not convey any useful information.
Since the oxidation of the amalgamated cobalt in carbonate buffer is
irreversible, reoxidation of the species did not occur and no peak appeared
in the anodic scan. Instead, another useful, but hardly used, EC technique,
electrochemical enrichment (EE), specifically devised for irreversible
redox couples has been employed.10 This
latter method makes use of three stages: preconcentration of the metal by
reduction at a potential more negative than the half-wave potential under
convective transport; stepping of the potential to a value more positive
than the half-wave potential in static solution so that the metal is
oxidised and the resulting ions accumulate at the electrode/solution
interface; and finally, a rapid scan to the cathodic direction to reduce
the ions generated in the previous stage. The reduction peak generated in
this final stage is a measure of the amount of accumulated metal. The
sequence of operations for the EE of Co(II) is illustrated in
Fig. 3(A). A typical reduction peak for
Co(II) after EE is shown in Fig.
3(B). It was found that, in a carbonate buffer with pH 10.2, the
cobalt reduction peak occurred at −1.34 V (Fig.
3(B)). The relationship between the current and the
Co(II) concentration was linear, indicating a promising
pre-separation behaviour.
Ep will be reduced and accumulated on
the electrode while metal ions with redox potential E
⩽
Ep will be unaffected. As soon as the potential is
stepped to a more positive stripping potential, Es, the
accumulated metals with redox potentials, Ei, where
Es > Ei >
Ep will be oxidised and stripped back into the solution
while those metals with redox potentials Ei >
Es will remain on the electrode. Thus, from the
potentially useful range (Eal, Ecl)
only the metals in the narrow range (Es,
Ep) will be available for detection. In this mode of
stripping, amperometric, spectroscopic or chemiluminescent detection can be
employed. For the present work, CL detection was selected due to its
simplicity and sensitivity. Dynamic EC techniques were initially used to
study the pre-separation behaviour of Co(II) on a mercury
electrode. The EC accumulation and stripping of Co(II) was
investigated on a hanging mercury drop electrode (HMDE). ASV was initially
investigated but this technique did not convey any useful information.
Since the oxidation of the amalgamated cobalt in carbonate buffer is
irreversible, reoxidation of the species did not occur and no peak appeared
in the anodic scan. Instead, another useful, but hardly used, EC technique,
electrochemical enrichment (EE), specifically devised for irreversible
redox couples has been employed.10 This
latter method makes use of three stages: preconcentration of the metal by
reduction at a potential more negative than the half-wave potential under
convective transport; stepping of the potential to a value more positive
than the half-wave potential in static solution so that the metal is
oxidised and the resulting ions accumulate at the electrode/solution
interface; and finally, a rapid scan to the cathodic direction to reduce
the ions generated in the previous stage. The reduction peak generated in
this final stage is a measure of the amount of accumulated metal. The
sequence of operations for the EE of Co(II) is illustrated in
Fig. 3(A). A typical reduction peak for
Co(II) after EE is shown in Fig.
3(B). It was found that, in a carbonate buffer with pH 10.2, the
cobalt reduction peak occurred at −1.34 V (Fig.
3(B)). The relationship between the current and the
Co(II) concentration was linear, indicating a promising
pre-separation behaviour.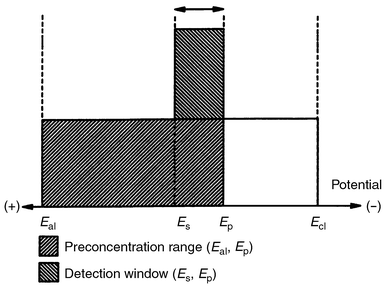 |
| | Fig. 2 Schematic diagram of the selectivity afforded by EC pre-separation. The
double arrow defines the detection window. | |
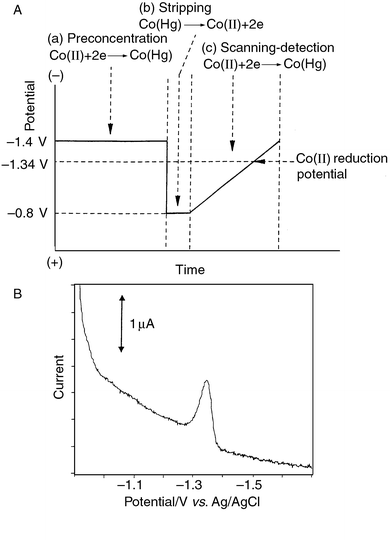 |
| | Fig. 3 (A) The potential–time profile employed in the technique of EE of
Co(II) and (B) A typical voltammetric response for 10 mg
l−1 Co(II) after EE on a HMDE. Potentials as in
Fig. 3(A); supporting electrolyte: carbonate
buffer pH 10.2; preconcentration time 120 s; stripping time 10 s; scan rate
100 mV s−1. | |
Choice of electrode and stability of the MFE
In a flow system, a MFE is preferable to a HMDE as the working electrode
due to its better mechanical stability, wide scope for various cell
configurations and large surface area.7
Compared to bare metal electrodes (like the gold ones employed in previous
work5,6), MFEs are easier to clean,
have a larger hydrogen overpotential (so that they can be safely used at
more cathodic potentials) and promote the reversibility of EC reactions. EC
regeneration and cleaning of the electrode from residual metals was
conducted electrochemically in the flowing carrier solution by holding the
potential of the electrode at an anodic value. An important advantage of
the MFE as opposed to the metal electrode reported earlier5,6 is that replacement of the electrode
surface after prolonged use is easier and more efficient and can be carried
out by simply stripping the mercury film at + 0.8 V and plating a new film
in situ. An important aspect of this work is that metals ions with
irreversible reduction kinetics (e.g. Co(II),
Ni(II), Mn(II)), undetectable by direct ASV, can be
quantified using the proposed method. The stability of the mercury film was
satisfactory under the conditions prevailing during the experiments. The
same mercury film could be used for 2–3 h without loss of activity
provided that the potential of the electrode was controlled to values more
cathodic than +0.2 V. Thus, the MFE was considered as the most appropriate
electrode for the particular application.Linearity, limit of detection and comparison between FIA
and continuous flow
Fig. 4(A) illustrates typical CL response
traces obtained for Co(II) standards in the range 100–700
μg l−1 after FIA EC pre-separation (3 injections). For
quantitative work, it was advantageous to use peak heights rather than peak
areas, especially for continuous flow pre-separation where tailing effects
are more pronounced, for reasons that will be discussed later. The
calibration parameters for both FIA and continuous flow pre-separations are
shown in Table 1. As expected,
continuous flow pre-separation offered higher sensitivity than FIA
pre-separation as already discussed in conjunction with stripping
voltammetry.7 The sensitivity and linear
range were, also, dependent on the accumulation time: for longer
pre-separation times the sensitivity increased but the linear range was
narrower. This property could be exploited to good effect in order to
tailor the sensitivity for different samples in which the concentration of
Co(II) is highly variable. The loss of linearity at higher
concentrations may be due to a combination of electrode overloading
(saturation) in the stage of metal accumulation or light quenching during
the CL detection. The limit of detection could be further improved by
extending the pre-separation period.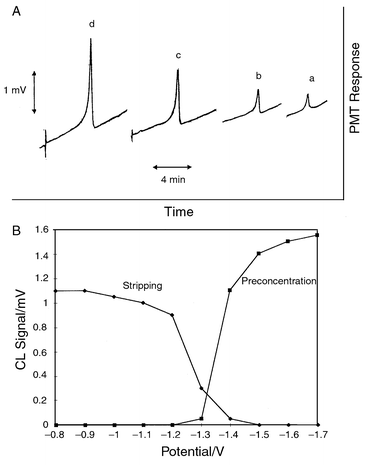 |
| | Fig. 4 (A) CL response for Co(II) after pre-separation on a MFE in
the FIA mode (3 injections). Traces (a), (b), (c), (d) represent
Co(II) concentrations of 100, 200, 400 and 700 μg
l−1, respectively. Potentials as in Fig. 3(A); (B) CL signal of a 400 μg
l−1 Co(II) solution as a function of: (i) the
pre-separation potential (■) when the stripping potential was set at
−0.8 V, and; (ii) the stripping potential (◆) when the
pre-separation was carried out at −1.4 V. Pre-separation in the FIA
mode (3 injections). | |
Table 1 Calibration parameters for the determination of Co(II) using
different pre-separation schemes and times
| Pre-separation mode | Upper limit of linear range/μg
l−1 | Sensitivity/ mV L μg−1 | Limit of detection/ μg l−1) (at the
3σ level) |
|---|
| FIA (3 injections) | 1 × 103 | 7 × 10−3 | 5 |
| Continuous flow (2 min) | 3 × 102 | 2 × 10−2 | 1.5 |
| Continuous flow (4 min) | 1 × 102 | 5 × 10−2 | 0.5 |
Selection of potentials and selectivity
The selection of the pre-separation and stripping potentials is of great
significance for the successful application of this method. As mentioned
earlier, the pre-separation potential must be set to a value more cathodic
than the redox half-wave potential of the metal of interest. The variation
of the CL signal as a function of the pre-separation potential is
illustrated in Fig. 4(B). Although more
cathodic potentials resulted in higher CL signals, in the case of
Co(II), a pre-separation potential of −1.4 V was selected
as a precaution to avoid hydrogen evolution which can occur at more
negative potentials. The selection of the stripping potential was equally
critical. The stripping potential should be set to a value more anodic than
the redox half-wave potential of the metal of interest. Fig. 4(B) illustrates the variation of the peak
height as a function of the stripping potential and suggests that the more
cathodic the stripping potential the lower the sensitivity of the CL
detection. The possible explanation of this effect is that a substantial
anodic overpotential is required to activate irreversible reactions (in
this case the oxidation of elemental cobalt). The more anodic the stripping
potential, the higher the anodic overpotential and the more efficient the
oxidation of cobalt. Nevertheless, excessively anodic stripping potentials
might compromise the selectivity of the method since the detection window
expands and other coexisting metals could strip off the electrode and
interfere with the analysis. To demonstrate the effect of the stripping
potential on the selectivity, a solution containing Co(II) and
Cu(II) was studied with the pre-separation potential set to
−1.4 V. The presence of Cu(II) did not affect the CL peak
heights when the stripping was carried out at −0.8 V since at this
potential only Co oxidised; however, when the stripping potential was set
to +0.2 V, oxidation of the pre-concentrated Cu occurred and the CL peak
height increased by more than 100% due to the contribution of the
Cu(II) ions to the total response. In cases where several metal
ions are present in the sample, the detection window should be as narrow as
possible to avoid interference from those metals that possess redox
potentials similar to that of cobalt. It was found that, in real samples,
it was more appropriate to use a stripping potential of −1.2 V for
Co(II) (i.e. marginally more anodic than the cobalt
reduction peak illustrated in Fig. 3(B)).
This narrow detection window of Es = −1.2 V,
Ep = −1.4 V, combined with the fact that, among
metal ions, Co(II) has the strongest catalytic activity on the
oxidation of luminol by hydrogen peroxide, endows the method with
satisfactory selectivity. Under these conditions, the following metal ions
did not interfere in the analysis of Co(II) at a 1000-fold
excess over Co(II): Zn(II), Cu(II),
Cd(II), Fe(III), Pb(II),
Cr(VI), Sn(IV), Ti(IV), Mg(II),
Mn(II). Ni(II) only interfered at concentrations over
200-fold higher than those of Co(II).Dispersion effects
The choice of the pre-separation mode has two serious implications in
terms of both sensitivity and shape of the response. A comparison of the
final CL signals was made for 60 μg l−1 Co(II) in the direct CL detection (i.e. without
pre-separation), CL detection after continuous flow pre-separation and CL
detection after FIA pre-separation. In particular, the ratio of peak
heights was 23∶8∶1 and the ratio of peak widths at half peak
heights was 1∶5∶2 for direct CL detection, CL detection after
continuous flow pre-separation for 4 min and CL detection after FIA
pre-separation. An interesting property of the detection is that the peak
heights after pre-separation are lower and wider than in direct CL
detection. This is due to two reasons. (i) The low coulometric efficiency
of the EC cell employed in this work that results in a low preconcentration
efficiency. A calculation based on a model derived previously in this
laboratory showed that less than 10% of the amount of Co(II)
flowing through the cell is reduced and accumulated on the MFE. Under
optimised conditions of flow rates and EC cell geometry, a coulometric
efficiency of 50% would be feasible. (ii) Dispersion effects which are more
pronounced in the pre-separation modes than in direct CL detection. The
physical presence of the EC cell causes an increase in dispersion. In
addition, the EC stripping process contributes to the dispersion as, upon
application of the stripping potential, oxidation of the accumulated Co is
neither instantaneous nor quantitative. Indeed, some of the oxidised Co
diffuses deep into the mercury bulk and needs some finite time to diffuse
back through the mercury film and out into the solution as an ion. These
ions will trail the main bulk of the stripped metal and cause tailing of
the CL peak. On the other hand, the dispersion in the continuous mode is
higher than in the FIA mode, as indicated by the ratio of the peak widths
at half peak heights and also made apparent by the pronounced tailing of
the peaks . This effect is due to the fact that the EC cell is further from
the detector in the continuous configuration than in the FIA configuration
(Fig. 1), meaning that the dispersion is
higher in the continuous flow configuration.Analytical application
The proposed method was applied to the analysis of a high purity iron
sample (149/3 BCR) with a certified cobalt content of 0.0073 ±
0.0005%. Since the cobalt concentration in the sample was relatively high,
the pre-separation was in the FIA mode. The cobalt content calculated using
our method was 0.0078 ± 0.0006% (n = 6) at the
3σ level. The experimentally determined mean value is higher
than the certified value and this is probably due to interference from the
presence of tartrate ions added to complex iron. Experiments with
artificial solutions indicated that, at concentration ratios similar to
that in the sample, tartrate causes a 5% error in the determination of
cobalt. Nevertheless, taking into consideration the complexity and the
composition of the sample, there is satisfactory statistical agreement
between the certified and determined content. Acknowledgements
The financial contribution of the Engineering and Physical Sciences
Research Council through a ROPA grant to AE is gratefully acknowledged.References
- U. Isacsson and G. Wettermark, Anal. Chim. Acta, 1974, 68, 339 CrossRef CAS.
- D. B. Paul, Talanta, 1978, 25, 377 CrossRef CAS.
- K. Robards and P. J. Worsfold, Anal. Chim. Acta, 1992, 266, 147 CrossRef CAS.
- Q. Lin, A. Guiraum and R. Escobar, Anal. Chim. Acta, 1993, 283, 379 CrossRef CAS.
- C. X. Zhang, S. C. Zhang and Z. J. Zhang, Analyst, 1998, 123, 1383 RSC.
- W. Qin, Z. J. Zhang and H. J. Liu, Anal. Chem., 1998, 70, 3579 CrossRef CAS.
- A. Economou, P. R. Fielden and A. J. Packham, Analyst, 1994, 119, 279 RSC.
- A. Economou and P. R. Fielden, Analyst, 1993, 118, 47 RSC.
- J. Wang,
Stripping Analysis, VCH,
Deerfield Beach, FL, 1985. Search PubMed.
- V. Stara and M. Kopanica, Electroanalysis, 1993, 5, 595 CAS.
Footnote |
| † Present address: Laboratory of Analytical Chemistry,
Department of Chemistry, Aristotle University of Thessaloniki, Thessaloniki
540 06, Greece. |
|
| This journal is © The Royal Society of Chemistry 2001 |
Click here to see how this site uses Cookies. View our privacy policy here.