Anomalous Schmidt reaction products of phenylacetic acid and derivatives
Carolyn C. Woodroofe, Boyu Zhong, Xingliang Lu and Richard B. Silverman*
Department of Chemistry, Northwestern University, Evanston, Illinois, 60208-3113, USA
First published on UnassignedUnassigned23rd December 1999
Abstract
Treatment of carboxylic acids with sodium azide in sulfuric acid normally results in decarboxylation with conversion of the carboxylic acid to an amine (the Schmidt reaction). However, many side reactions have been reported to occur, particularly in the case of α-aryl carboxylic acids, such as sulfonation, direct amination of the phenyl ring, cyclization to a lactam, and elimination of side chains to give aniline. In this study, the reactions of a variety of analogues of phenylacetic acid under given reaction conditions are examined to determine which characteristics are important in the competing side reactions. Some reactions were carried out with TEMPO free radical as a radical scavenger to investigate whether direct amination proceeds by a radical intermediate. Phenylacetic acid is shown to give an ortho-aminated diamine product instead of the para-aminated one expected from direct amination. A mechanism for this side reaction, involving cyclization to a lactam intermediate followed by further cleavage, is proposed; an analogue of the hypothetical intermediate has been isolated for biphenylacetic acids.
Introduction
In the classic Schmidt reaction, the acid-catalyzed addition of hydrazoic acid to an organic acid replaces the carboxy group with an amine moiety.1,2 The alkyl group R bonded to the carboxylic acid undergoes a migration from the original carbonyl carbon to the neighboring nitrogen of the azide moiety, eliminating N2 and later CO2 (Scheme 1). However, the Schmidt reaction of aromatic carboxylic acids has produced a few unexpected results over the years. For example, both sulfonation of the phenyl ring and cyclization to give a lactam were reported by Datta et al.3 in γ-phenylbutyric acid and δ-phenylvaleric acid (Scheme 2), and elimination of alkyl side chains to give aniline as the major amine product in a variety of α-arylcarboxylic acids was reported by Palmere![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 4 (Scheme 3).
4 (Scheme 3).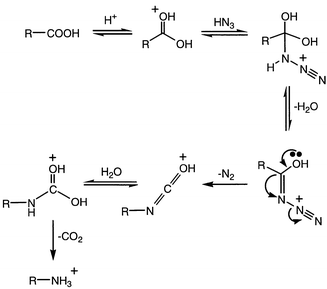 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
Here we report the effects of slight differences in structure on competing side reactions of the Schmidt reaction of phenylacetic acid and derivatives to try to rationalize the anomalous behavior.
Results and discussion
When phenylacetic acid (1, R = H, Scheme 4) was subjected to standard conditions of the Schmidt reaction, the expected benzylamine product was obtained, as well as o-aminobenzylamine (5). Since electron donation of aromatic systems in lieu of alkyl migration is well documented,3–6 a mechanism consistent with electrophilic aromatic substitution can be proposed (Scheme 4). The reaction begins as in the usual Schmidt reaction (Scheme 1) to give 2.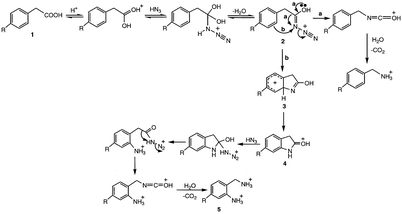 | ||
| Scheme 4 | ||
Schmidt rearrangement (pathway a) with loss of N2 gives the isocyanate, which undergoes hydrolysis with decarboxylation to benzylamine. Alternatively, however (pathway b), intramolecular electrophilic aromatic substitution leads to 3; deprotonation produces protonated oxoindole (4). Further reaction with azide leads to o-aminobenzylamine (5). To test this mechanism two experiments were carried out. In one, the reaction was run with only 1.5 equiv. of sodium azide, but no neutral product (4) was isolated. In a second experiment, oxoindole was treated with azide and sulfuric acid under the same conditions as was phenylacetic acid. Complete decomposition of the oxoindole occurred, but no amines could be found. These experiments suggest that 4 is not a true intermediate in the mechanism, but possibly a hydrate or other closely-related derivative is an active intermediate, which is not able to form from oxoindole itself prior to decomposition of the oxoindole. Several other aryl- and α-arylcarboxylic acids were investigated (Table 1), but formation of the corresponding ortho-amine was not duplicated by any other acid tested.
| Carboxylic acid | Normal Schmidt product | p-Amine | o-Amine | Sulfonate |
|---|---|---|---|---|
| 1 (R = NO2) | 100 | |||
| 1 (R = Cl) | 99 | 1 | ||
| 1 (R = H) | 77 | 23 | ||
| 1 (R = CH3) | 8 | 1 | 91 | |
| 10 | 77 | 23 |
The effectiveness of pathway b in Scheme 4 relies on the electron-donating ability of the phenyl ring; therefore, m-tolylacetic acid (1, R = CH3), m-chlorophenylacetic acid (1, R = Cl), and m-nitrophenylacetic acid (1, R = NO2) were investigated as phenylacetic acid analogues to determine the ratio of normal Schmidt reaction to the side pathway via intramolecular electrophilic aromatic substitution. The results, shown in Table 1, are consistent with electrophilic aromatic substitution, even though only the parent compound produced the o-amino-substituted benzylamine. Whereas phenylacetic acid gives 77% of the normal Schmidt product, increasing the electron withdrawing substitution gives increasing amounts of this product; when R = Cl, 99% is formed, and when R = NO2, 100% is obtained. The electron-rich tolyl analogue gives mostly electrophilic aromatic substitution as a result of sulfation of the ring (6 and 7), not intramolecular amination, but only 8% of the normal Schmidt product (8) and 1% of the diamine (9, Scheme 5).
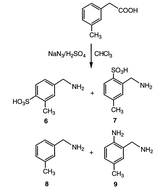 | ||
| Scheme 5 | ||
1-Phenylcyclopropanecarboxylic acid (10) also was studied in the Schmidt reaction, and it was found to produce 77% of the normal Schmidt product and 23% of the para-substituted diamine (11).
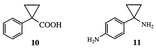
Direct amination of benzene rings by hydrazoic acid was observed by Schmidt in 19247 and was suggested by Keller and Smith8 to proceed by a radical mechanism. However, reactions of toluene under Schmidt conditions gave predominantly the para-amine with no trace of o-toluidine, so it is not clear if a radical reaction is important. To test whether a radical mechanism is involved in the para-amination of 1-phenylcyclopropanecarboxylic acid (10), the reaction was also conducted in the presence of the radical scavenger TEMPO. As summarized in Table 2, the yield of the diamine product 11 in this reaction decreased, but did not disappear until the molar concentration of TEMPO was more than twice that of the sodium azide. The yield of 11 decreased with increasing amounts of radical scavenger, but the yield of normal Schmidt product remained relatively constant. These results suggest that the para-amination reaction is the result of a radical reaction, and, because of the amount of radical scavenger required, the reaction is not a chain reaction. As suggested by Schmidt,7 this reaction probably proceeds by the generation of a triplet nitrene intermediate, which inserts into the least hindered of the C–H bonds of the substrate.
| Reagent equivalents | Product yield (%) | |||
|---|---|---|---|---|
| TEMPO | 10 | NaN3 | Schmidt product | 11 |
| 0 | 1 | 2.8 | 77 | 23 |
| 1 | 1 | 2.8 | 89 | 11 |
| 5.9 | 1 | 2.8 | 100 | 0 |
To gain evidence for an intramolecular ortho-amination of phenylacetic acid, leading to the o-aminobenzylamine (5, Scheme 4) via a lactam intermediate, biphenyl-2-carboxylic acid (12, R = H; Scheme 6), which should form a stable lactam intermediate, was subjected to the conditions of the Schmidt reaction. No diaminated product was observed, but instead the corresponding tricyclic lactam, phenanthridinone (16 or 18, R = H), the expected intermediate in the formation of the corresponding ortho-amino compound, was obtained (Scheme 6). Two mechanisms are drawn in Scheme 6 for the formation of this product from intermediate 13. Pathway a is similar to the mechanism in Scheme 4 for ortho-amination of phenylacetic acid. Aryl migration gives 14, which undergoes electrophilic aromatic substitution to 15; loss of a proton gives phenanthridinone (16). Alternatively, (pathway b) electrophilic aromatic substitution at nitrogen gives 17; loss of a proton produces 18, which, when R = H, is identical to 16. The normal Schmidt reaction is shown in pathway c from 14, yielding 19, which was not observed. A radical mechanism was excluded by running the reaction in the presence of TEMPO free radical, which did not inhibit the formation of phenanthridinone.
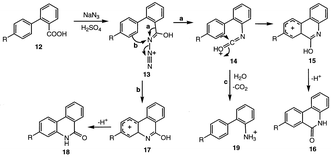 | ||
| Scheme 6 | ||
In an attempt to differentiate pathways a and b in Scheme 6, the same reaction was run with 4′-(trifluoromethyl)biphenyl-2-carboxylic acid (12, R = CF3). However, different products were obtained depending on the length of time of reaction. After 3 hours of reflux, a 1∶1 mixture of the normal Schmidt product (19, R = CF3) and the diamine (19, R = NH2, trapped as the Boc derivative) was observed. However, after 15 h of reflux none of the normal Schmidt product (19, R = CF3) was detected. Instead, four new compounds were isolated, 16/18 (R = CF3) and 16/18 (R = COOH), as well as the diamine (19, R = NH2) in a ratio of 21∶5∶35%, respectively. Unfortunately, lactams 16/18 (R = CF3) could not be cleanly separated nor could 16/18 (R = COOH), but the trifluoromethyl lactams were obtained in about four times the amount as the corresponding carboxyl lactams. Not knowing which structure corresponds to the major isomer, it is not definite that the mechanism in Scheme 6 is operative, but, based on Scheme 6, the major isomer should be 16 (R = CF3 or COOH); the electron-withdrawing properties of these substituents should favor pathway a in Scheme 6 over pathway b. The carboxylate substituent probably derives from an initial hydrolysis of the trifluoromethyl group, a reaction known to occur in sulfuric acid.9 The diamine product (19, R = NH2) is, most likely, derived from a hydrolysis of the trifluoromethyl group to the carboxylic acid, which is converted, along with the other carboxylic acid, via a normal Schmidt reaction, to the diamine.
These results indicate that small changes in the structures of aromatic-containing carboxylic acids can have a large effect on the direction of the Schmidt reaction. Generation of highly electrophilic species leads to potential intramolecular electrophilic aromatic substitution reactions, which produce the major side products.
Experimental
Solvents were obtained from Fisher Chemical Co. and reagents were obtained from Aldrich Chemical Co.; all were used without further purification. NMR spectra were obtained on a 300 MHz Gemini-300 spectrometer in CDCl3 unless otherwise specified. Melting points were obtained on a Fisher-Johns apparatus and are uncorrected. Elemental microanalyses were performed by Oneida Research Services (Whiteboro, NY).General Schmidt reaction with phenylacetic acid
Phenylacetic acid (1.91 g, 14 mmol) was dissolved in 75 mL of CHCl3 in a 250 mL 3-necked round bottom flask equipped with a stir bar, rubber stoppers, and a reflux condenser sealed with a septum. Concentrated sulfuric acid (10 mL, 98%) was added to the stirred solution, and the mixture was heated in an 80 °C oil bath. Sodium azide (3.4 g, 52 mmol) was added portionwise, and the mixture was allowed to reflux for 3 h. The mixture was removed from the heat, quenched with 50 mL of ice water, and stirred an additional 15 min, then poured into a separatory funnel, and the layers were separated. The aqueous layer was diluted to 200 mL. The organic layer was extracted with 2 × 25 mL of 1.0 M HCl, and the combined aqueous layers were washed with 2 × 30 mL of ether, basified with 6 M NaOH, cooled and extracted with 4 × 50 mL of ethyl acetate. The ethyl acetate solution was washed with 2 × 50 mL of saturated NaCl solution, dried over Na2SO4, and evaporated to give 1.0 g of a yellow oil. The oil was dissolved in 24 mL of methanol and 6 mL of triethylamine in a 100 mL round bottom flask, and di-tert-butyl dicarbonate (3.27 g, 15 mmol) was added. The solution was stirred overnight at room temperature; then the triethylamine and methanol were removed by evaporation on the rotary evaporator. The carbamate mixture was separated on a 15 cm × 20 mm silica gel column, eluting with CH2Cl2. Two products were recovered:Benzylamine tert-butyl carbamate (1.0 g, 30%), was recrystallized from hexane to give the product as chunky white crystals; mp 52–53 °C (lit.10 53–54 °C); 1H NMR δ 1.49 (9 H, s), 4.32 (2 H, d), 4.87 (1 H, br s), 7.3 (5 H, m); 13C NMR δ 28.5, 44.7, 79.5, 127.4, 127.5, 128.6, 139, 156; HRMS Calcd. for C12H17NO2-CH3 192.1024. Found 192.1025.
o-Aminobenzylamine tert-butyl dicarbamate (5 di-tert-butyl dicarbamate) was recrystallized from hexane (405 mg, 9%); mp 125–126.5 °C; 1H NMR δ 1.48 (9 H, s), 1.53 (9 H, s), 4.28 (2 H, d), 4.9 (1 H, br s), 7.0 (1 H, t), 7.14 (1 H, d), 7.29 (1 H, t), 8.0 (2 H, m); 13C NMR δ 28, 42, 121, 128, 130; HRMS Calcd. for C17H26N2O4 322.189. Found 322.189; Anal. Calcd. C, 63.33; H, 8.13; N, 8.69. Found C, 63.21; H, 8.18; N, 8.49%.
Reaction of m-chlorophenylacetic acid (1, R = Cl)
m-Chlorophenylacetic acid (1, R = Cl; 2.39 g, 14 mmol) was subjected to the same reaction and extraction conditions as in the General Schmidt Reaction, yielding 1.56 g of a brown oil. This was stirred with 6.3 g of di-tert-butyl dicarbonate in 20% triethylamine in methanol at room temperature overnight, and after the solvent was evaporated, the resulting residue was separated on a silica gel column (17 cm × 40 mm, 80 g silica), eluting with 4∶1 hexane–ethyl acetate. Two products were isolated:m-Chlorobenzylamine tert-butyl carbamate, which was recrystallized from hexane to give iridescent white crystals (1.74 g, 51%); mp 47–48 °C; 1H NMR δ 1.47 (9 H, s), 4.3 (2 H, d), 4.9 (1 H, br s), 7.17 (1 H, s), 7.26 (3 H, m); 13C NMR δ 28, 44, 103, 125, 128, 130. Anal. Calcd. for C12H16ClNO2, C, 59.63; H, 6.67; N, 5.80; Cl, 14.67. Found C, 59.56; H, 6.66; N, 5.85; Cl, 14.48%.
4-Amino-3-chlorobenzylamine tert-butyl carbamate; 23.3 mg (0.6%) after recrystallization from hexane; mp 71–73 °C; 1H NMR δ 1.47 (s, 9 H), 4.2 (d, 2 H), 4.8 (br s, 1 H), 6.74 (d, 1 H), 7.0 (d, 1 H), 7.2 (s, 1 H); 13C NMR δ 28.4, 44, 116, 119, 127, 129, 130, 142, 156. Anal. Calcd. for C12H17ClN2O2, C, 56.14; H, 6.67; N, 10.91. Found C, 56.40; H, 6.62; N, 10.72%.
Reaction of m-nitrophenylacetic acid (1, R = NO2)
m-Nitrophenylacetic acid (1, R = NO2; 1.67 g, 9.2 mmol) gave a dark brown oil (1.23 g, 89%). The amine was protected as the tert-butyl carbamate by stirring overnight at room temperature with di-tert-butyl dicarbamate (8.85 g, 41 mmol) in 20% triethylamine in methanol. The solvent was evaporated to give a creamy solid (2.53 g), which was purified on a silica gel column (25 g, 17 cm × 30 mm), eluting with 4∶1 hexane–ethyl acetate. Recrystallization from hexane gave m-nitrobenzylamine tert-butyl carbamate (1.63 g, 70%); mp 75–76 °C; 1H NMR δ 1.48 (9 H, s), 4.43 (2 H, d), 5.08 (1 H, br s), 7.52 (1 H, t), 7.64 (1 H, d), 8.14 (2 H, m); 13C NMR δ 28.4, 43.8, 80, 103.4, 122, 122.3, 130, 133.4, 141.5, 148.4, 156. Anal. Calcd. for C12H16N2O4, C, 57.13; H, 6.39; N, 11.10. Found C, 57.17; H, 6.36; N, 11.02%.Reaction of m-tolylacetic acid (1, R = CH3)
m-Tolylacetic acid (2.10 g, 14 mmol) was allowed to react as described for phenylacetic acid. Upon quenching with water, white crystals of m-tolylbenzylamine sulfate salt, sulfonated at the 4 and 6 positions (6 and 7), began to form within 15 min of stirring. This product was recrystallized from methanol to give flaky white crystals (1.70 g, 60%); mp (decomp.) > 285 °C; 1H NMR (D2O) δ 2.35 (3 H, s), 4.4 (2 H, s), 7.35 (2 H, m), 7.77 (1 H, d); 13C NMR δ 20, 41.4, 127.6, 129, 130.3, 139, 143.4, 198.1; Anal. Calcd. for C8H11NO3S, C, 47.75; H, 5.48; N, 6.95; S, 15.95. Found C, 47.70; H, 5.51; N, 6.92; S, 15.90%. The organic extracts of the aqueous layer were worked up as described for phenylacetic acid, and two products were obtained:m-Methylbenzylamine (8) tert-butyl carbamate (152 mg, 5%); mp 55–56 °C; 1H NMR (CDCl3) δ 1.47 (s, 9 H), 2.45 (s, 3 H), 4.30 (d, 2 H), 4.91 (br, 1 H), 7.17 (s, 1 H), 7.26 (m, 3 H).
2-Amino-5-methylbenzylamine (9) tert-butyl carbamate (30 mg, 0.9%); mp 122–124 °C; 1H NMR (CDCl3) δ 1.47 (s, 9 H), 1.53 (s, 9 H), 2.47 (s, 3 H), 4.21 (d, 2 H), 4.83 (br, 2 H), 6.74 (s, 1 H), 7.02 (m, 2 H).
Reaction of 1-phenylcyclopropanecarboxylic acid (10)
1-Phenylcyclopropanecarboxylic acid (10, 1.49 g, 9.2 mmol) gave 0.70 g of a bright yellow oil. The oil was stirred with di-tert-butyl dicarbonate (4.4 g, 20 mmol) in a solution of 20% triethylamine in methanol at room temperature overnight. The solvent was removed on a rotary evaporator, and the residue was dissolved in CHCl3, adsorbed onto a small amount of silica gel, and separated on a silica gel column (18 cm × 20 mm, 20 g silica gel), eluting with 6∶1 hexane–ethyl acetate. Two products were recovered:The standard Schmidt product, 1-phenylcyclopropylamine tert-butyl carbamate (0.315 g, 16%); mp 77–78 °C (from hexane); 1H NMR δ 1.27 (4 H, m), 1.41 (9 H, s), 5.25 (1 H, br s), 7.23 (5 H, m); 13C NMR δ 18, 28, 36, 80, 125 (2), 126, 128, 144, 155; Anal. Calcd. for C14H19NO2, C, 72.07; H, 8.21; N, 6.00. Found C, 71.97; H, 8.23; N, 6.02%.
1-(4-Aminophenyl)cyclopropylamine (11) di-tert-butyl dicarbamate (0.154 g, 5%); mp 171–173 °C (from hexane); 1H NMR δ 1.2 (4 H, m), 1.3 (9 H, s), 1.5 (9 H, s), 5.3 (1 H, br s), 6.55 (1 H, br s), 7.15 (2 H, d), 7.25 (2 H, d); 13C NMR δ 17.8, 28.3, 79.5, 80.5, 118, 128, 136, 138, 153, 155; Anal. Calcd. for C19H28N2O4, C, 65.49; H, 8.10; N, 8.04. Found C, 65.51; H, 8.13; N, 8.04%.
Reaction of 1-phenylcyclopropanecarboxylic acid (10) with TEMPO
The general procedure above was followed except that 1.4 g (9.2 mmol) of TEMPO free radical was added prior to the addition of the sodium azide. The dark red liquid resulting from the workup was stirred with di-tert-butyl dicarbonate (6.6 g, 30 mmol) in 20% triethylamine in methanol overnight. The residue was separated on a silica gel column (40 mm × 18 cm, 70 g silica gel), eluting first with 9∶1 hexane–ethyl acetate, then with 6∶1 hexane–ethyl acetate. The crude 1-phenylcyclopropylamine tert-butyl carbamate (0.546 g, 27%) was recrystallized from hexane to give 0.320 g (16%). The recrystallized yield of 11 di-tert-butyl dicarbamate was 66 mg (2%).Reaction of 1-phenylcyclopropanecarboxylic acid with increased TEMPO
The procedure above was followed with the exception that 8.20 g of TEMPO (54 mmol) was added prior to addition of the sodium azide. The extraction gave 6.80 g of a red liquid product, which was stirred with di-tert-butyl dicarbonate (5 g, 23 mmol) and passed through a silica gel column (14 cm × 40 mm, 60 g silica), eluting with 9∶1 hexane–ethyl acetate. The yellow-white crude product (0.70 g, 33%) was recrystallized from hexane to give 0.278 g (13%) of 1-phenylcyclopropylamine tert-butyl carbamate. No 11 di-tert-butyl dicarbamate was detected.Reaction of biphenyl-2-carboxylic acid (12, R = H)
Biphenyl-2-carboxylic acid (12, R = H; 0.91 g, 4.6 mmol) was dissolved in chloroform (25 mL) and 8 mL of concentrated sulfuric acid (98%) and was allowed to react with sodium azide (1.13 g, 17.4 mmol). Quenching with 50 mL of ice water gave a cream-colored suspension, which was filtered by suction, and the resulting solid was washed sequentially with deionized H2O, 1 M NaOH, and diethyl ether, and dried by suction to give the product as a beige powder (0.79 g, 84%). The product was recrystallized from acetone to give pure phenanthridinone (16 (R = H), 0.45 g, 50%); mp 288–290 °C (lit.11 293 °C). 1H NMR (DMSO) δ 7.20 (1 H, t), 7.28 (1 H, d), 7.40 (1 H, d), 7.47 (1 H, t), 7.85 (1 H, t), 8.30 (1 H, d), 8.33 (1 H, d), 8.50 (1 H, d), 11.70 (1 H, br s); 13C NMR (DMSO) δ 116, 118, 122, 122.5, 123, 126, 127.5, 128, 129.5, 133, 134, 136, 161. HRMS Calcd. for C13H9NO, 195.0670. Found 195.0672. Chloroform was added to the filtrate, and the remaining reaction mixture was worked up as in the reaction of phenylacetic acid, but no amine was recovered.Reaction of biphenyl-2-carboxylic acid (12, R = H) with TEMPO
The same reaction as above was carried out in the presence of TEMPO (2.83 g, 18.1 mmol). After quenching as above, 1.13 g yellow-brown solid was filtered. A ninhydrin test of the filtrate indicated that no free amine was present. Recrystallization from acetone gave 0.57 g (64%) of phenanthridinone (16, R = H).Reaction of 4′-trifluoromethylbiphenyl-2-carboxylic acid (12, R = CF3)
4′-Trifluorobiphenyl-2-carboxylic acid (1.23 g, 4.6 mmol) was dissolved in 35 mL of CHCl3 and was stirred at room temperature. Concentrated sulfuric acid (8 mL) was added, and the mixture was heated to reflux for 1.5 h. Sodium azide (1.13 g, 17.4 mmol) was added portionwise, then the reaction was worked up as described for phenylacetic acid. Evaporation of the solvent in vacuo gave a yellow oil, which was dissolved in aqueous 3 M HCl and evaporated to dryness. The residue was recrystallized from ethanol–ether to give powdery crystals of 19 (R = CF3) (377 mg, 22%); mp 155–157 °C; 1H NMR (CDCl3) δ 7.4 (t, 1 H), 7.5 (m, 3 H), 7.6 (q, 3 H), 10.1 (br, 3 H); 13C NMR (CDCl3) δ 122, 124,1 126, 128, 129, 130, 130.5, 131, 131.5, 136, 140; Anal. Calcd. for C13H11ClF3N, C, 57.05; H, 4.05; N, 5.12. Found C, 57.12; H, 4.03; N, 4.95%; HRMS Calcd. for C13H11ClF3N 237.227. Found 237.225.The aqueous layers were worked up as usual to give a brown oil (0.50 g). The oil was stirred with di-tert-butyl dicarbamate (2.18 g, 10 mmol) in 24 mL of methanol and 6 mL of triethylamine overnight at room temperature. The product was purified on a silica gel column, eluting with 3∶1 hexane–ethyl acetate to give powdery biphenyl 2-amino-4′-tert-butyl carbamate (the free base of 19, R = BocNH; 285 mg, 22%); mp 140–141 °C; 1H NMR (CDCl3) δ 1.55 (s, 9 H), 3.75 (br, 2 H), 6.5 (br, 1 H), 6.8 (m, 2 H), 7.1 (m, 2 H), 7.4 (q, 4 H); 13C NMR CDCl3δ 28.4, 81.0, 115.6, 118.7, 119, 127, 128.4, 129.7, 130.5, 134, 137.5, 143.7, 153; Anal. Calcd. for C17H20N2O2, C, 71.81; H, 7.09; N, 9.85. Found C, 71.80; H, 7.07; N, 9.79%; HRMS Calcd. for C17H20N2O2 284.361. Found 284.358.
In a second experiment, the reaction time was extended to 15 h with heating at reflux. The product from the aqueous layer was the same as at the 1.5 h reflux experiment, but the crude product after evaporation of the organic layer was different as analyzed by HPLC on a C18 column, eluting with 0.06% TFA in acetonitrile and water (50∶50) at 254 nm and a flow rate of 1 mL min−1. Two kinds of products were isolated by preparative HPLC, but each product contained two isomers, as evidenced by NMR spectroscopy. The components also were determined to be isomers by elemental analysis and MS of the mixtures.
2-Trifluoromethylphenanthridinone (16 and 18, R = CF3; 254 mg, 21%).
Mp 245–255 °C; 1H NMR (DMSO-d6) δ 7.2–7.9 (m, 4 H), 8.2–8.7 (m, 3 H), 11.9 (s, 1 H); 19F NMR (DMSO-d6) δ −61.46, −61.56; Anal. Calcd. for C14H8F3NO, C, 63.85; H, 3.06; N, 5.32. Found C, 63.45; H, 3.20; N, 5.15%; LC-MS: (M + H) 264, EI-MS 263.
2-Carboxyphenanthridinone (16 and 18, R = COOH; 40 mg, 5%).
Mp > 260 °C; 1H NMR (DMSO-d6) δ 7.2–8.0 (m, 4 H); 8.3–8.8 (m, 3 H); 11.8 (s, 1 H); 19F NMR (DMSO-d6) δ no fluorine peak was observed; Anal. Calcd. for C14H9NO3, C, 70.29; H, 3.79; 5.86. Found C, 69.81; H, 4.05; N, 5.63%; LC-MS: (M + H) 240.
The aqueous layer was worked up as usual to give the free base of 19 (R = BocNH); 454 mg, 35%.
Acknowledgements
The authors are grateful for financial support for this work from the U.S. Public Health Service, National Institutes of Health (GM32634).References
- J. March , Advanced Organic Chemistry: Reactions, Mechanisms and Structures, 4th edn., Wiley, New York, 1992, pp. 1093–1095. Search PubMed.
- M. S. Newman and H. L. Gildenhorn, J. Am. Chem. Soc., 1948, 70, 317 CrossRef CAS.
- S. K. Datta, C. Grundmann and N. K. Bhattacharyya, J. Chem. Soc., 1970, 2058 Search PubMed.
- R. M. Palmere and R. T. Conley, J. Org. Chem., 1970, 35, 2703 CrossRef CAS.
- R. Faure, J. Heterocycl. Chem., 1994, 31, 141 Search PubMed.
- G. Di Maio and V. Permutti, Tetrahedron, 1966, 22, 2059 CrossRef.
- K. F. Schmidt, Ber. Dtsch. Chem. Ges., 1924, 57, 704 Search PubMed.
- R. N. Keller and P. A. S. Smith, J. Am. Chem. Soc., 1944, 66, 1122 CrossRef CAS.
- M. A. McClinton and D. A. McClinton, Tetrahedron, 1992, 48, 6555 CrossRef CAS.
- H. E. Baumgarten, H. L. Smith and A. Staklis, J. Org. Chem., 1975, 40, 3554 CrossRef CAS.
- T. Paterson, R. Smalley and H. Suschitzky, Synthesis, 1975, 709 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |