Peculiar reactivity of face to face biscorrole and porphyrin–corrole with a nickel(II) salt. X-Ray structural characterization of a new nickel(II) bisoxocorrole
François
Jérôme
a,
Jean-Michel
Barbe
a,
Claude P.
Gros
a,
Roger
Guilard
*a,
Jean
Fischer
b and
Raymond
Weiss
*b
aUni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersité de Bourgogne, LIMSAG UMR 5633, Faculté des Sciences Gabriel, 6, bouleard Gabriel, 21100-, Dijon, France. E-mail: rguilard@u-bourgogne.fr; Fax: (33) 0380396117
ersité de Bourgogne, LIMSAG UMR 5633, Faculté des Sciences Gabriel, 6, bouleard Gabriel, 21100-, Dijon, France. E-mail: rguilard@u-bourgogne.fr; Fax: (33) 0380396117
bUniersité Louis Pasteur, Institut Le Bel, 4, rue Blaise Pascal, 67000, Strasbourg, France. E-mail: fischer@chimie.u-strasbg.fr; Fax: (33) 0388415363
First published on 4th December 2000
Abstract
New nickel(II) bisoxocorrole complexes have been synthesized and characterized. Two nickel bisoxocorrole isomer complexes are formed in a 1:1 ratio when a biscorrole, where an anthracenyl bridge links the two macrocycles in a face to face arrangement, is metallated by nickel in air. Evidence for the insertion of an oxo group at one of the meso positions is given by the X-ray structural characterization of the Ni2(BOCA) “cis” isomer where BOCA is a bisoxocorrole with an anthracenyl spacer. Conversely, under the same reaction conditions no nickel bisoxocorrole is obtained when biphenylene is the linker of the biscorrole system, the major product of the reaction being a bisnickel bisradical species. Only a small amount of a bisnickel hydrooxobiscorrole is isolated in this reaction. A reaction mechanism for both series (anthracenyl and biphenylenyl) is given and a comparison of the behavior of the biscorrole system with related derivatives is proposed.
Introduction
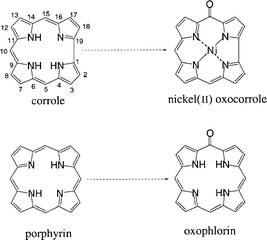
Numerous researches have been devoted to the molecular structure, spectroscopy and electronic properties of metallocorroles.1–5 Although nickel complexes were among the first metallocorroles reported,6 their structure was not clearly defined for a long time.6–11 Indeed, until 1997, the structure of the nickel corrole was uncertain due to its paramagnetic nature.8,11–15 Furthermore, the low intensity of the Soret band led the authors to conclude that the corrole complex was not totally aromatic and they postulated a nickel(II) complex with an “extra” hydrogen atom at one of the three meso positions. In 1997 the electrochemistry data16 and a crystal structure determination17 demonstrated that no such hydrogen atom was present at this position. Conversely a distorted square planar geometry indicated an interruption of the corrole ring aromaticity. Magnetic measurements and the unusual low intensity of the double band in the Soret region of the electronic spectrum were in accordance with a radical nature for the nickel complex.1,17 The authors pointed out that the formal nickel(III) complex was in fact a nickel(II) one with a delocalized electron in the π orbital of the ligand. However, they assumed that the nickel(II) radical came from the loss of an hydrogen radical from an intermediate nickel(II) complex with a free imine not coordinated to the nickel.13 This intermediate was in equilibrium by an hydrogen radical migration with a nickel(II) complex with an “extra” hydrogen atom at one of the meso positions.We recently reported the synthesis of four “ face to face” bismacrocycles containing either two corrole rings5 or a porphyrin and a corrole unit,4 rigidly linked by two different bridges such as anthracenyl and biphenylenyl spacers. Here, we show an interesting behaviour of these bismacrocycles when metallated with a nickel salt. Indeed, the insertion of nickel into these “face to face” bismacrocycles leads to the formation of a novel family of macrocycles, named “bisoxocorroles ”, similar to the oxophlorins observed in porphyrin chemistry (Scheme 1).18–22
 | ||
| Scheme 1 | ||
These bisoxocorroles are characterized by the presence of an oxygen atom at one of the meso positions, precisely at the C-15 and C-15′ or C-5′ positions (′ is given for the second macrocycle). So far to our knowledge, this is the first example of such a bismacrocycle ever described. The synthesis, the X-ray characterization and 1H NMR studies of the nickel bisoxocorrole Ni2(BOCA) (BOCA is a bisoxocorrole with an anthracenyl spacer) as well as related derivatives are reported in this article. A probable mechanism for this peculiar reaction is given and reference is made to the different behaviour of monomeric nickel corroles.
Results
Synthesis
A standard procedure was employed to metallate the anthracenyl bridged biscorrole 1 with nickel(II). The biscorrole was heated at 80°C in pyridine in the presence of an excess of nickel(II) acetate under air, the metallation reaction being monitored by UV-visible spectroscopy. After purification, the 1H NMR and mass spectrometry data led us to postulate the formation of a nickel bisoxocorrole. In contrast to monomeric corroles, the formation of radical species was not observed and the isolated compounds were found diamagnetic indicating the presence of low spin nickel(II). The formation of two bisoxocorrole isomers 2a and 2b occurred in 38% yield, the oxygen atoms being either on the same side of the bismacrocycle (2a) or on opposite sides (2b) (Scheme 2). | ||
| Scheme 2 | ||
The two bisoxocorrole isomers were easily separated by column chromatography on silica gel. 2b was first collected indicating that isomer 2b was less polar than 2a. Both isomers were isolated in the same quantity (19% yield). Mass spectrometry measurements indicated a single ion at m/z 1688 ([M]+•) for both derivatives in accordance with the presence of two oxygen atoms on the bismacrocycle.
Contrary to the anthracenyl derivative, using the same metallation reaction conditions, the formation of a bisoxocorrole was not observed in the case of the biphenylenyl derivative 3, the major product being the bisradical species 4 along with a small amount of a mixed-ligand complex 5. Spectroscopic data of 5 were consistent with the presence of both oxocorrole and hydrocorrole moieties. Mass spectrometry analysis confirmed the probable structure of 5 with a peak at m/z 1648 compared to 1632 for the bisradical 4 (Scheme 3).
 | ||
| Scheme 3 | ||
Characterization of the nickel bisoxocorroles 2a and 2b
Nickel bisoxocorroles were characterized by 1H NMR, UV-visible, infrared, mass spectrometry and by X-ray crystallography.During the metallation reaction, a typical change in the UV-visible spectrum is observed (Table 1). A decrease in intensity of the Soret band is noted which indicates a breakdown of the aromaticity in the macroring. For instance, the ε value for the Soret band of the anthracenyl biscorrole free base 1 is 1.31 × 105 whereas ε is close to 0.80 × 105 (dm3 mol−1 cm−1) for the nickel bisoxocorroles 2a and 2b. Moreover, the Soret band is extremely broad due to the superposition of three bands between 379 and 416 nm. A single Q band appears at 635 nm with a low ε value of ca. 8.0 × 103 dm3 mol−1 cm−1 (Fig. 1). It is noteworthy that the shape of the UV-visible spectra of 2a and 2b is nearly identical to that of the monomeric nickel corroles.
| λ max/nm (ε/10−3 dm3 mol−1 cm−1) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Spacer | Compound | Solvent | Soret band | Q bands | ||||
| a Relative intensity. b TMEC = 7,8,12,13,-tetraethyl-2,3,17,18-tetramethylcorrolato. | ||||||||
| Anthracenyl | 1 | CH2Cl2 | — | 416 | — | — | 572 | 608 |
| (131.5) | (35.4) | (29.0) | ||||||
| 7 | Pyridine | — | 417 | — | 524 | 582 | 605 | |
| (100.0)a | (27.3)a | (29.9)a | (33.3)a | |||||
| 7 | CH2Cl2 | — | 420 | — | 546 | 578 | — | |
| (100.0)a | (26.5)a | (26.5)a | ||||||
| 2a | CH2Cl2 | 379 | 391 | 411 | — | — | 635 | |
| (82.4) | (84.3) | (75.6) | (7.5) | |||||
| 2b | CH2Cl2 | 380 | 394 | 416 | — | — | 635 | |
| (78.7) | (85.3) | (79.2) | (8.5) | |||||
| 12 | CH2Cl2 | 402 | — | — | 524 | 558 | — | |
| (170.2) | (30.1) | (34.2) | ||||||
| Biphenylenyl | 3 | CH2Cl2 | — | 404 | — | — | 578 | 607 |
| (129.0) | (39.5) | (31.0) | ||||||
| 4 | Pyridine | — | 420 | — | 517 | 591 | 614 | |
| (79.4) | (44.7) | (37.6) | (43.4) | |||||
| 4 | CH2Cl2 | — | 417 | — | 543 | 571 | — | |
| (73.1) | (37.5) | (30.7) | ||||||
| 5 | CH2Cl2 | 350 | 386 | 420 | — | — | 672 | |
| (60.3) | (62.4) | (58.3) | (6.8) | |||||
| — | Ni(TMEC)17b | CH2Cl2 | 358 | 383 | 479 | 529 | 595 | 653 |
| (62.2) | (48.9) | (5.4) | (3.2) | (3.3) | (11.0) | |||
 | ||
| Fig. 1 UV-visible spectra of H6(BCA) and Ni2(BOCA) species. | ||
2a and 2b are diamagnetic indicating the lack of any radical species and the presence of a pure low spin nickel(II) complex. 1H NMR allows one to locate the oxygen atom since only one proton is present at the meso positions. Moreover, the disruption of the macrocycle aromaticity decreases the ring current and therefore the signal of the remaining meso proton of the oxocorrole moiety appears around δ 4 compared to 9 for the starting biscorrole 1 (See Table 2 for resonances of meso protons and Experimental Section for δ of other proton sites). Furthermore, the 1H NMR spectra of 2a and 2b clearly show the dissymmetry of the molecule (absence of C2 symmetry) which confirms the presence of only one oxygen atom at one of the two available meso positions. For instance, one signal is observed for the four methyl groups of the symmetric biscorrole 1 compared to two signals at δ − 0.22 and − 0.19 for those of the bisoxocorrole 2b.
| δ H meso | |||||
|---|---|---|---|---|---|
| Spacer | Compound | H-5 | H-15 | H-5′ | H-15′ |
| Anthracenyl | 1 | 8.86 | 8.86 | 8.86 | 8.86 |
| 2a | — | 3.93 | — | 3.93 | |
| 2b | — | 4.05 | 4.05 | — | |
| 12 | — | 2.87 | — | — | |
| Biphenylenyl | 3 | 9.59 | 9.59 | 9.59 | 9.59 |
| 5 | 4.02(H-5 or 5′) | 3.90(2H) | 4.01(H-5 or 5′) | — | |
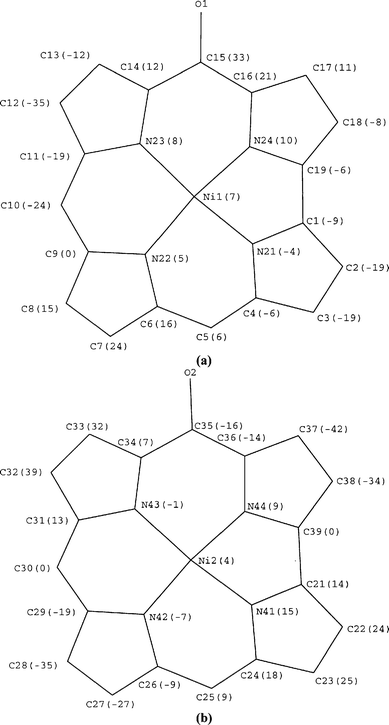
Side- and top-views of the molecular structure of Ni2(BOCA) 2a are displayed in Fig. 2(a) and 2(b), respectively, with the labelling scheme used for all the non-hydrogen atoms. Fig. 3(a) and 3(b) show, respectively, in Å × 10−2 units the perpendicular displacements of the nickel and the oxocorrole core atoms relative to the oxocorrole mean planes. Fig. 4 illustrates the orientation and position of the mean planes of the oxocorrole rings relative to that of the anthracenyl spacer. Table 3 lists selected bond distances and angles as well as some averages data for Ni2(BOCA)·2CH2Cl2·2H2O.
 | ||
| Fig. 2 (a) Side- and (b) top-views of the molecular structure and atom numbering of Ni2(BOCA). Thermal ellipsoids enclose 30% probability. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 3 Perpendicular displacements in Å × 10−2 units of (a) Ni1 and the Ni1 oxocorrole-core atoms and (b) Ni2 and the Ni2 oxocorrole-core atoms relative to the corresponding oxocorrole mean planes. | ||
 | ||
| Fig. 4 Orientation and position of the oxocorrole ring mean planes relative to the anthracenyl spacer. | ||
| Ni1–N21 | 1.861(6) | Ni2–N44 | 1.858(6) |
| Ni1–N22 | 1.880(6) | 〈Ni–Np〉 | 1.871(13) |
| Ni1–N23 | 1.894(6) | 〈Np–Cα〉 | 1.371(20) |
| Ni1–N24 | 1.859(6) | 〈Cα–Cβ〉 | 1.431(25) |
| Ni2–N41 | 1.859(6) | 〈Cβ–Cβ〉 | 1.381(19) |
| Ni2–N42 | 1.873(6) | 〈Cm–Cα〉 | 1.417(36) |
| Ni2–N43 | 1.880(6) | 〈Cα–Cα〉 | 1.441(7) |
| 〈Np–Cα–Cβ〉 | 109.3(9) | 〈Cα–Cm–Cα〉 | 123.8(9) |
| 〈Cα–Np–Cβ〉 | 107.5(9) | 〈Np–Cα–Cα〉 | 110.8(9) |
| 〈Cα–Cβ–Cβ〉 | 106.8(9) |
As shown in Fig. 2(a) and 2(b), both nickel atoms, Ni1 and
Ni2, lie in a four-coordinate distorted square-planar environment.
The average Ni1–Np and Ni2–Np bond distances are not
significantly different, their mean value being 1.871(6) Å. The shortest Ni1–Np and Ni2–Np bond distances, Ni1–Np(24) 1.859(6) and Ni1–Np(21) 1.861(6), Ni2–Np(41) 1.859(5) and Ni2–Np(44) 1.858(6)
Å, involve pyrrole nitrogens belonging to
rings linked directly
![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia the Cα–Cα bond. Similar features have been observed previously in the
structures of other metallocorroles.1,2 The average Ni–Np
bond
distance in 2a is slightly longer than that of 1.844 Å in the
nickel(II) derivative of the tetramethyltetraethylcorrole π-radical
cation, NiII(TMEC•+), in which the nickel(II) cation lies also in
a distorted square-planar environment.17
However, the Ni–Np
bond
distances present in Ni2(BOCA) are shorter than the corresponding
ones observed for the large number of reported
structures of nickel(II) porphyrins,23 the averages lying
between 1.888 Å in Ni(TCHP) and 1.960 Å in Ni(DeutDME) (TCHP = tetracyclohexylporphyrinate and DeutDME = 2,4-diacetyldeuteroporphyrinate dimethyl ester).23 This in 2a
can be
ascribed to the reduced hole size of the two oxocorrole rings
occurring in [BOCA]4− relative to the hole size of the porphyrin
core. A similar decrease of the M–Np
bond lengths is observed
in several metallocorroles as different as
[Rh(OMC)(AsPh3)], [Co(OMTPC)(PPh3)] and Mn(OMC) (OMC = 2,3,7,8,12,13,17,18-octamethylcorrolate;
OMTPC =
2,3,7,8,12,13,17,18-octamethyl-5,10,15-triphenylcorrolate).24–26
ia the Cα–Cα bond. Similar features have been observed previously in the
structures of other metallocorroles.1,2 The average Ni–Np
bond
distance in 2a is slightly longer than that of 1.844 Å in the
nickel(II) derivative of the tetramethyltetraethylcorrole π-radical
cation, NiII(TMEC•+), in which the nickel(II) cation lies also in
a distorted square-planar environment.17
However, the Ni–Np
bond
distances present in Ni2(BOCA) are shorter than the corresponding
ones observed for the large number of reported
structures of nickel(II) porphyrins,23 the averages lying
between 1.888 Å in Ni(TCHP) and 1.960 Å in Ni(DeutDME) (TCHP = tetracyclohexylporphyrinate and DeutDME = 2,4-diacetyldeuteroporphyrinate dimethyl ester).23 This in 2a
can be
ascribed to the reduced hole size of the two oxocorrole rings
occurring in [BOCA]4− relative to the hole size of the porphyrin
core. A similar decrease of the M–Np
bond lengths is observed
in several metallocorroles as different as
[Rh(OMC)(AsPh3)], [Co(OMTPC)(PPh3)] and Mn(OMC) (OMC = 2,3,7,8,12,13,17,18-octamethylcorrolate;
OMTPC =
2,3,7,8,12,13,17,18-octamethyl-5,10,15-triphenylcorrolate).24–26
As shown in Fig. 2(a) and 2(b), one oxygen atom is bonded
to a meso-carbon of each ring and only the cis isomer of Ni2(BOCA) is present in the studied crystals. The two meso-positions occupied by an oxygen atom are C15 and C35 giving two cis Cm![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O groups oriented in the same direction. The bond distances of 1.24(1) (C15–O1) and 1.26(1) Å (C35–O2) are in good agreement with expected CO bond lengths. CO bond distances of similar values have been observed in metallooxophlorin.27
The Cα–Cm bond distances adjacent to the CmO groups are not significantly different. Their mean value of 1.45(1) Å is slightly longer than the mean value of the four other Cα–Cm bond lengths of 1.40(1) Å of the two rings present in Ni2(BOCA), but similar to the known distance for a single Csp2–Csp2 bond (1.44 Å). Moreover, both CO groups are not coplanar with the corrole mean planes, the dihedral angles between the C14-C15(O1)-C16 and C34-C35(O2)-C36 mean planes and the oxocorrole mean planes of both rings being 12.8(2) and 14.0(2)°, respectively. These structural features indicate that only a slight delocalization of the CO bond occurs across the π system of the corrole ring.
O groups oriented in the same direction. The bond distances of 1.24(1) (C15–O1) and 1.26(1) Å (C35–O2) are in good agreement with expected CO bond lengths. CO bond distances of similar values have been observed in metallooxophlorin.27
The Cα–Cm bond distances adjacent to the CmO groups are not significantly different. Their mean value of 1.45(1) Å is slightly longer than the mean value of the four other Cα–Cm bond lengths of 1.40(1) Å of the two rings present in Ni2(BOCA), but similar to the known distance for a single Csp2–Csp2 bond (1.44 Å). Moreover, both CO groups are not coplanar with the corrole mean planes, the dihedral angles between the C14-C15(O1)-C16 and C34-C35(O2)-C36 mean planes and the oxocorrole mean planes of both rings being 12.8(2) and 14.0(2)°, respectively. These structural features indicate that only a slight delocalization of the CO bond occurs across the π system of the corrole ring.
Although the core of the corrole π-cation radical ligand in NiII(7,8-TMEC•+) is totally planar,17 the two rings of Ni2(BOCA) are slightly distorted. Both oxocorrole core conformations correspond to a mixture of irregular saddle and ruffle and doming distortions. The saddle distortions may be characterized by the out of plane displacements of the geminal β-pyrrole carbons of + 0.106(7) and − 0.213(7) Å for the Ni1- and + 0.302(7) and − 0.346(7) Å for the Ni2-containing macrocycles above and below the oxocorrole mean planes. The doming of both rings may be described by the separation between the 4Np mean plane and oxocorrole mean plane of each ring of 0.050(1) and 0.108(1) Å for the Ni1- and Ni2-containing macrocycles, respectively. The irregular ruffling may be characterized by the orientations of the Cβ–Cβ bonds relative to the oxocorrole mean plane of each ring, the corresponding angles having values close to 0 (C2–C3), 3.8(2) (C7–C8), 169.8(2) (C12–C13), 8.0(2) (C17–C18), 0(C22–C23), 176.9(2) (C27–C28), 3.3(2) (C32–C33) and 176.7(2)° (C37–C38).
The structure of Ni2(BOCA) exhibits features similar to those reported for the nickel(II) bisporphyrin complex Ni2(DPA) (DPA = diporphyrin anthracene) with the same rigid anthracenyl bridge.28,29 The oxocorrole moieties in Ni2(BOCA) are not stacked over one another, but are slightly slipped with respect to each other. A lateral shift of 1.535(2) Å occurs between Ni2 and the projection of Ni1 on the mean plane of the oxocorrole ring containing Ni2 (Fig. 4). This lateral shift leads to a Ni···Ni distance of 4.678(1) Å and a slip angle of 19.1(6)°. The distance between a virtual plane containing the meso-carbon C15 which is parallel to the mean plane of the Ni2 oxocorrole is 4.419(2) Å. The dihedral angle of this virtual plane and the mean plane of the Ni1 oxocorrole is 7.8(3)° (Fig. 4). In the case of the Pacman porphyrin, Ni2(DPA), the lateral shift is 2.40 Å giving a Ni···Ni distance of 4.566 Å and an interring separation of 3.88 Å.
The anthracenyl spacer is almost planar; the largest deviation from its mean plane is 0.073(7) Å (C80), the mean deviation 0.041(7) Å. The nickel(II) bisoxocorrole molecules, Ni2(BOCA), are stacked top to bottom in columns along the [011] crystal direction. The shortest intermolecular contact distances have values of 3.6 Å.
Spectroscopic characterization of the nickel hydrooxobiscorrole 5
The mixed-ligand nickel hydrooxobiscorrole complex 5 has been characterized by 1H NMR (see Experimental section). The meso methylene group of the hydrocorrole moiety is evidenced as a double doublet resonance at δ 3.90. Two singlets appear at δ 4.01 and 4.02 corresponding to the single meso protons of the hydrocorrole and oxocorrole macrocycles. Likewise the lack of C2 symmetry is enhanced by the presence of four different signals around δ 2.2 for the four methyl groups of the bismacrocycle. Interestingly, and in contrast to the anthracenyl derivative, only one isomer is detected by 1H NMR. Hydrooxobiscorroles and bisoxocorroles have nearly the same UV-visible spectra indicating loss of ring aromaticity and a large Soret band centered at 386 nm is observed for 5 (Table 1).Characterization of the bisradical species 7 and 4
In order better to understand the mechanism of formation of these bisoxocorroles and hydrooxobiscorrole, the same metallation reactions were carried out under an argon atmosphere to evaluate the role of dioxygen in this reaction. Under the same experimental conditions, but in the absence of dioxygen, we could isolate the bisradical nickel complex 4 (biphenylenyl spacer) and 7 (anthracenyl spacer) (Scheme 4). The UV-visible spectra are different from those described before for the bisoxocorrole, the hydrooxobiscorrole or the monomeric corrole nickel complexes (see Table 1). For example, in pyridine, the electronic absorption spectrum of 7 exhibits a Soret band at 417 nm and three Q bands at 524, 582 and 605 nm. A similar UV-visible spectrum is observed for 4 (Soret band 420 nm; Q bands 517, 591, 614 nm) (Table 1). Compound 7 is paramagnetic and very broad signals are observed on the 1H NMR spectrum between δ − 2 and 30 while for the biphenylenyl derivative 4 broad signals appear between δ 0 and 9. The ESR spectrum of 7 shows a characteristic signal of a radical delocalized on the macrocycle and split into three lines (g1 = 2.004, g2 = 2.007, g3 = 2.017) at 100 K as observed for monomeric nickel corroles.16,17 The ESR spectrum of 4 exhibits a very low intensity signal in the same region as for 7, thus indicating a stronger interaction between the two radicals in 4 than in 7. ESR and 1H NMR are together in accordance with the formulation as a nickel(II) bisradical for derivatives 4 and 7. Additional evidence supporting this hypothesis comes directly from the mass spectrum exhibiting a single ion at m/z 1658 for 7 and 1632 for 4. It should be noted that the bisradical species 7 is very unstable under dioxygen or air and decomposition occurs under these conditions only after a few minutes. Conversely, 4 is more stable and no extensive decomposition is observed after three days in solution. The direct oxygenation of these bisradical species does not lead to the formation of bisoxocorroles or mixed hydrooxobiscorroles respectively. This indicates that the bisradicals 4 and 7 are not the intermediates in the oxocorrole formation. | ||
| Scheme 4 | ||
Synthesis and characterization of the bisnickel porphyrin oxocorrole derivative 12
In order to dismiss an eventual chemical cooperativity between the two corrole macrocycles during the reaction, the porphyrin–corrole bismacrocycle 10 was metallated by a nickel salt. After three hours of reaction in pyridine under reflux only the corrole macrocycle was metallated as shown by mass spectrometry. Attempts to isolate this monometallated complex 11 were unsuccessful due to an extensive decomposition during the purification. This complex was only identified in the reaction medium by mass spectrometry by the presence of one peak at m/z 1353 corresponding to a nickel(II) oxocorrole face to face to a porphyrin free base. Taking into account the higher ε value for the porphyrin ring compared to corrole and oxocorrole ones, the nickel(II) oxocorrole–porphyrin free base 11 exhibits the same UV-visible spectrum as that of the starting porphyrin–corrole 10. Metallation of the porphyrin ring only occurs in benzonitrile under reflux leading to the formation of the bisnickel(II) derivative 12 (Scheme 5).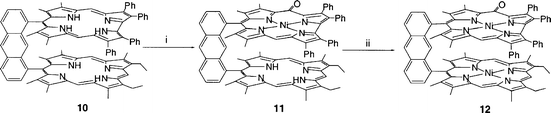 | ||
| Scheme 5 | ||
The mass spectrum of complex 12 consists in a single peak at m/z 1410 and the 1H NMR spectrum reveals the total dissymmetry. The meso proton of the oxocorrole moiety is shifted downfield (δ = 2.87) compared to that of the bisoxocorrole 2a or 2b (δ≈4) or the biscorrole (δ≈9) (see Table 2 and Experimental section). This results from the superposition of two phenomena. First, as in the case of bisoxocorroles, the ring current is lower than for monocorroles and therefore the signal appears downfield. Secondly, the addition of the strong ring current of the porphyrin over the oxocorrole moiety shifts this signal more downfield. It is interesting that this molecule is chiral and therefore two stereoisomers are formed during ring oxidation of the corrole.
Discussion
Proposed mechanism for formation of the bisoxocorroles 2a and 2b
From the results described, the characterization of the hydrooxobiscorrole 5, the difference in stability of the two bisradical species 4 and 7 in air, and the mechanism for metallation of corroles by nickel proposed in the literature, we can suggest a mechanism which corresponds to the physicochemical data obtained. A concerted mechanism implying a dioxygen molecule attack and a hydrogen radical migration from the imine to the meso position is proposed in Scheme 4.The first step is analogous to that described in the literature for the monomeric corrole series; it consists in the formation of a bisnickel(II) biscorrole 6 with one imine group of each corrole ring not co-ordinated. Under a dioxygen atmosphere, the migration of the hydrogen radical from the remaining imine group to the meso position concerted with a dioxygen molecule attack (8) generates the transient bishydroperoxide 9a. This unstable intermediate immediately loses two water molecules to give the corresponding bisoxocorrole 2a. This mechanism perfectly explains the formation of only one oxo group per corrole ring. The formation of the bisoxocorrole is only due to a radical species generated by insertion of the nickel into the cavity of the corrole. The access to isomer 2b can be explained by the same mechanism if one takes into account the possible delocalization of the radical on the second meso position of 6.
Proposed mechanism for the formation of complexes 4 and 5
A radical interaction can account for the difference in reactivity of compounds 1 and 3. As described for “Pacman” porphyrins30–36 or biscorroles5 and porphyrin–corroles,4 the spacer length (anthracenyl or biphenylenyl) is of major importance in the reactivity of the complex formed. For the anthracenyl derivative the two corrole rings behave as independent macrocycles since no interaction occurs due to the length of the bridge (4.9 Å). For the biphenylenyl analogue the shorter distance between the two corrole rings (3.8 Å) leads to a stronger interaction between the two macrocycles. Therefore, the major formation of the bisradical species 4 is observed and the hydrooxobiscorrole 5 is produced as a minor compound. Furthermore, no bisoxocorrole derivative, which is the main product in compound 1 metallation by nickel, is formed. The strong interaction between the two radicals in 4 is demonstrated by the 1H NMR spectrum which exhibits signals in a diamagnetic region. However, the spacer is not short enough to observe a complete antiferromagnetic coupling between the two radicals and this can explain the broadness of the NMR signals. The formation in low yield of only one hydrooxobiscorrole isomer follows from this interaction. Conversely, for the anthracenyl compound, the distance between the two corrole rings is too large to observe a spin interaction between the two radicals and each radical is delocalized on each meso position and statistically the two isomers 2a and 2b are generated in the same amount.It is worth pointing out that a monomeric corrole bearing a phenyl group at the C-10 position does not afford the nickel(II) oxocorrole but the nickel(II) radical. The formation of oxocorroles is observed only in the case of face to face bismacrocycles. This probably stems from a larger electron density at the meso position than at other carbon atoms, compared to the monomeric corroles, resulting from the superposition of the two corrole rings. Molecular modeling calculations currently underway will probably answer this question.
Finally, infrared spectra of all the derivatives were recorded
and typical values for the carbonyl group stretching bands of
bisoxocorroles were found. As in the case of oxophlorins
(1560–1580 cm−1)22 or dipyrroketones, the carbonyl vibration frequencies arise around 1600 cm−1 (see Experimental section). This low value for a carbonyl group vibration indicates a certain degree of polarization of the CO bond. In comparison with metallated oxophlorins, a weaker polarization is
found for the carbonyl group of the oxocorrole which seems to indicate a stronger delocalization of the CO group of the oxophlorins than that of the oxocorrole.22
Experimental
Instrumentation
IR spectra were recorded on a Bruker IFS 66v FTIR spectrophotometer for samples prepared as 1% dispersions in KBr pellets, UV-visible spectra on a Varian Cary 5 spectrophotometer. EPR measurements were performed on a Bruker ESP 300 instrument at 100 K in toluene. The g values were measured relative to diphenylpicrylhydrazyl (dpph) (g = 2.0037 ± 0.002). Basic alumina (Merck; usually Brockmann Grade III, i.e. deactivated with 6% water) and silica gel (Merck; 70–120 μm) were used for column chromatography. 1H NMR spectra were recorded on a Bruker AC 200 Fourier transform spectrometer of the “Centre de Spectrométrie Moléculaire de l’Université de Bourgogne”, chemical shifts are expressed in ppm relative to chloroform (7.258 ppm). Microanalyses were performed at the Université de Bourgogne on a Fisons EA 1108 CHNS instrument. Mass spectra were obtained on Bruker ProFLEX III MALDI/TOF (matrix assisted laser desorption ionization time of flight) spectrometer mode using dithranol as matrix. X-Ray diffraction data were collected on a Enraf-Nonius Kappa-CCD diffractometer (Mo-Kα radiation, λ = 0.71073 Å).Chemicals
For all syntheses, chemicals were commercially available and used without further purification. Syntheses of face to face biscorroles and porphyrin–corroles were performed using a previously described procedure.4,5Syntheses
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2960 (CH), 2930 (CH), 2857 (CH) and 1633
cm−1 (CO); Calc. for C112H82N8Ni2O2·(CH3OH) C 78.85, H 5.04, N 6.51; found C 78.72, H 4.99, N 6.63%. 2b: 1H NMR (200 MHz, CDCl3, 25°C) δ
− 0.22 (s, 6H, CH3),
− 0.19 (s, 6H, CH3), 0.31 (t, 6H, CH2CH3), 0.39 (t, 6H, CH2CH3), 1.48 (m, 8H, CH2CH3), 4.05 (s, 2H, meso-CH), 6.08–7.81 (m, 46H, Ph, anthracene), 8.21 (s, 1H, anthracene) and 8.83 (s, 1H,
anthracene); MS (MALDI/TOF) m/z
= 1688 ([M]+•); IR (KBr)
= 2956 (CH), 2930 (CH), 2860 (CH) and 1631 cm−1 (CO); calc. for C112H82N8Ni2O2·CH3OH. C 78.85, H 5.04, N 6.51; found C 79.05, H 4.95, N 6.47%.
= 3068 (CH), 3034 (CH), 2961 (CH), 2925 (CH), 2852 (CH) and 1624 cm−1
(CO) Calc. for C55H41N4NiO: C 80.11, H 5.01, N 6.79. Found: C 79.93, H 5.11, N 6.83%.
= 2962 (CH), 2926
(CH), 2861 (CH) and 1621 cm−1 (CO). Calc. for C91H72N8Ni2O·(H2O):
C 76.49, H 5.22, N 7.84. Found: C 76.25, H 5.43, N 7.23%.
= 2960 (CH), 2930 (CH), 2857 (CH) and 1633
cm−1 (CO); Calc. for C112H82N8Ni2O2·(CH3OH) C 78.85, H 5.04, N 6.51; found C 78.72, H 4.99, N 6.63%. 2b: 1H NMR (200 MHz, CDCl3, 25°C) δ
− 0.22 (s, 6H, CH3),
− 0.19 (s, 6H, CH3), 0.31 (t, 6H, CH2CH3), 0.39 (t, 6H, CH2CH3), 1.48 (m, 8H, CH2CH3), 4.05 (s, 2H, meso-CH), 6.08–7.81 (m, 46H, Ph, anthracene), 8.21 (s, 1H, anthracene) and 8.83 (s, 1H,
anthracene); MS (MALDI/TOF) m/z
= 1688 ([M]+•); IR (KBr)
= 2956 (CH), 2930 (CH), 2860 (CH) and 1631 cm−1 (CO); calc. for C112H82N8Ni2O2·CH3OH. C 78.85, H 5.04, N 6.51; found C 79.05, H 4.95, N 6.47%.
= 3068 (CH), 3034 (CH), 2961 (CH), 2925 (CH), 2852 (CH) and 1624 cm−1
(CO) Calc. for C55H41N4NiO: C 80.11, H 5.01, N 6.79. Found: C 79.93, H 5.11, N 6.83%.
= 2962 (CH), 2926
(CH), 2861 (CH) and 1621 cm−1 (CO). Calc. for C91H72N8Ni2O·(H2O):
C 76.49, H 5.22, N 7.84. Found: C 76.25, H 5.43, N 7.23%.
Crystal structure determination of complex 2a
Suitable single crystals of Ni2(BOCA)·2CH2Cl2·2H2O 2a were obtained by slow evaporation of a dichloromethane–methanol solution at room temperature. All experimental parameters used are given in Table 4. For all subsequent calculations the Enraf-Nonius OpenMoleN package was used.37 The structure was solved using direct methods. Hydrogen atoms were introduced as fixed contributors in structure factor calculations by their computed coordinates (C–H 0.95 Å) and isotropic thermal parameters such as B(H) = 1.3 Beqv(C) Å2 but not refined. Water and CH2Cl2 protons were omitted. One of the CH2Cl2 and one of the water molecules are disordered over 2 sites in the ratio 1:1. Full least-squares refinements on F. A final difference map revealed no significant maxima. The scattering factor coefficients and anomalous dispersion coefficients come respectively from Tables 2.2b and 2.3.1 of ref. 38.| Formula | C112H82N8Ni2O2·2CH2Cl2·2H2O |
| Molecular weight | 1895.27 |
| Crystal system | Triclinic |
| Space group | P1 |
| a/Å | 16.1351(7) |
| b/Å | 16.3746(6) |
| c/Å | 19.6653(7) |
| α/° | 109.763(3) |
| β/° | 93.411(3) |
| γ/° | 90.830(3) |
| U/Å3 | 4877.7(7) |
| Z | 2 |
| μ/mm−1 | 0.554 |
| T/K | 173 |
| Number of data measured | 37354 |
| Number of data with | 8937 |
| I>3σ(I) | |
| R | 0.073 |
| Rw | 0.107 |
CCDC reference number 440/223. See http://www.rsc.org/suppdata/nj/b0/b007623f/ for crystallographic files in .cif format.
Acknowledgements
We are grateful to the French Ministry of Research (MENRT) and CNRS (UMR 5633) for financial support. The “ Région Bourgogne” and “ Air Liquide” are acknowledged for scholarships (FJ). The authors are also grateful to M. Soustelle for assistance in the synthesis of pyrrole precursors.References
- C. Erben, S. Will and K. M. Kadish, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 2, p. 233. Search PubMed.
- R. Paolesse, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 2, p. 201. Search PubMed.
- C. Tardieux, C. P. Gros and R. Guilard, J. Heterocycl. Chem., 1998, 35, 965 Search PubMed.
- F. Jérôme, C. P. Gros, C. Tardieux, J.-M. Barbe and R. Guilard, New J. Chem., 1998, 22, 1327 RSC.
- F. Jérôme, C. P. Gros, C. Tardieux, J.-M. Barbe and R. Guilard, Chem. Commun., 1998, 2007 RSC.
- A. W. Johnson and I. T. Kay, J. Chem. Soc., 1965, 1620 RSC.
- R. Grigg, A. W. Johnson and G. Shelton, Liebigs Ann. Chem., 1971, 32, 746 Search PubMed.
- Y. Murakami, Y. Matsuda, K. Sakata, S. Yamada, Y. Tanaka and Y. Aoyama, Bull. Chem. Soc. Jpn., 1981, 54, 163 CAS.
- I. D. Dicker, R. Grigg, A. W. Johnson, H. Pinnock, K. Richardson and P. Van den Broek, J. Chem. Soc. C, 1971, 536 RSC.
- R. Grigg, A. W. Johnson and G. Shelton, J. Chem. Soc. C, 1971, 2287 RSC.
- N. S. Hush, J. M. Dyke, M. L. Williams and I. S. Woolsey, J. Chem. Soc., Dalton Trans., 1974, 395 RSC.
- A. W. Johnson, in Porphyrins and Metalloporphyrins, ed. K. M. Smith, Elsevier, Amsterdam, 1975, p. 729. Search PubMed.
- R. Grigg, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 2, p. 327. Search PubMed.
- N. S. Hush and M. L. Dyke, J. Inorg. Nucl. Chem., 1973, 35, 4341 Search PubMed.
- H. R. Harrison, O. J. R. Hodder and D. Crowfoot Hodgkin, J. Chem. Soc. B, 1971, 640 RSC.
- K. M. Kadish, V. A. Adamian, E. Van Caemelbecke, E. Gueletii, S. Will, C. Erben and E. Vogel, J. Am. Chem. Soc., 1998, 120, 11986 CrossRef CAS.
- S. Will, J. Lex, E. Vogel, H. Schmickler, J. P. Gisselbrecht, C. Haubtmann, M. Bernard and M. Gross, Angew. Chem., Int. Ed. Engl., 1997, 36, 357 CrossRef CAS.
- M. D. G. H. Vicente, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 2, p. 149. Search PubMed.
- R. G. Khoury, L. Jaquinod, A. M. Shachter, N. Y. Nelson and K. M. Smith, Chem. Commun., 1997, 215 RSC.
- R. G. Khoury, L. Jaquinod, R. Paolesse and K. M. Smith, Tetrahedron, 1999, 55, 6713 CrossRef CAS.
- R. G. Khoury, L. Jaquinod, D. J. Nurco, R. K. Pandey, M. O. Senge and K. M. Smith, Angew. Chem., Int. Ed. Engl., 1996, 35, 2496 CrossRef CAS.
- P. S. Clezy, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 2, p. 103. Search PubMed.
- W. R. Scheidt, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 3, p. 49. Search PubMed.
- R. Paolesse, S. Licoccia, G. Bandoli, A. Dolmella and T. Boschi, Inorg. Chem., 1994, 33, 1171 CrossRef CAS.
- T. Boschi, S. Licoccia, R. Paolesse, P. Tagliatesta, M. Azarnia Tehran, G. Pelizzi and F. Vitali, J. Chem. Soc., Dalton Trans., 1990, 463 RSC.
- S. Licoccia, E. Morgante, R. Paolesse, F. Polizio, M. O. Senge, E. Tondello and T. Boschi, Inorg. Chem., 1997, 36, 1564 CrossRef CAS.
- A. L. Balch, Coord. Chem. Rev., 2000, 200–202, 349 CrossRef CAS.
- J. P. Collman, P. S. Wagenknecht and J. E. Hutchison, Angew Chem. Int. Ed., 1994, 33, 1537 CrossRef.
- J. P. Fillers, K. G. Ravichandran, I. Abdalmuhdi, A. Tulinsky and C. K. Chang, J. Am. Chem. Soc., 1986, 108, 417 CrossRef CAS.
- M. Lachkar, A. Tabard, S. Brandès, R. Guilard, A. Atmani, A. De Cian, J. Fischer and R. Weiss, Inorg. Chem., 1997, 36, 4141 CrossRef CAS.
- R. Guilard, M. A. Lopez, A. Tabard, P. Richard, C. Lecomte, S. Brandès, J. E. Hutchison and J. P. Collman, J. Am. Chem. Soc., 1992, 114, 9877 CrossRef CAS.
- R. Guilard, S. Brandès, A. Tabard, N. Bouhmaida, C. Lecomte, P. Richard and J. M. Latour, J. Am. Chem. Soc., 1994, 116, 10202 CrossRef CAS.
- J. P. Collman, P. S. Wagenknecht, J. E. Hutchison, N. S. Lewis, M. A. Lopez, R. Guilard, M. Lher, A. A. Bothnerby and P. K. Mishra, J. Am. Chem. Soc., 1993, 115, 4947 CrossRef CAS.
- J. P. Collman, Y. Y. Ha, P. S. Wagenknecht, M. A. Lopez and R. Guilard, J. Am. Chem. Soc., 1993, 115, 9080 CrossRef CAS.
- J. P. Collman, Y. Y. Ha, R. Guilard and M. A. Lopez, Inorg. Chem., 1993, 32, 1788 CrossRef CAS.
- J.-M. Barbe and R. Guilard, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 2, p. 211. Search PubMed.
- OpenMoleN, Interactive Structure Solution, Nonius B. V., Delft, 1997..
- D. T. Cromer and J. T. Waber, International Tables for X-Ray Crystallography, Kynoch Press, Birmingham, 1974, vol. 4. Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |