Highly distorted d8-rhodium(+I) and d10-rhodium(−I) complexes: synthesis, reactivity, and structures in solution and solid state
Stephan Deblon, Heinz Rüegger, Hartmut Schönberg, Sandra Loss, Volker Gramlich and Hansjörg Grützmacher*
Laboratory of Inorganic Chemistry, ETH-Centre, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersitätstrasse 6, CH-8092, Zürich, Switzerland. E-mail: gruetzmacher@inorg.chem.ethz.ch; Fax: + 41 1 632 10 90
ersitätstrasse 6, CH-8092, Zürich, Switzerland. E-mail: gruetzmacher@inorg.chem.ethz.ch; Fax: + 41 1 632 10 90
First published on 23rd October 2000
Abstract
A synthesis of the new ligand 5-diphenylphosphanyl-10-methyl-5H-dibenzo[a,d]cycloheptene (metroppph) was developed in order to prepare highly distorted tetra-co-ordinated rhodium(+I) complexes. The ligand metroppph contains a cycloheptatriene ring in a rigid boat conformation such that a Ph2P and an olefinic binding site are perfectly arranged for transition metal complexation. Four equivalents of metroppph react with [Rh2(μ-Cl)2(cod)2] in the presence of KPF6 to yield almost quantitatively [Rh(+I)(metroppph)2]PF6, which was isolated in the form of dark red-violet crystals. A crystal structure analysis reveals that the co-ordination sphere of the rhodium centre in [Rh(+I)(metroppph)2]+ deviates strongly from a square planar arrangement (φ = 42°). One methyl group in [Rh(+I)(metroppph)2]+ can be deprotonated by KOBut to give the allyl complex [Rh(allyltroppph)(metroppph)]. This complex has a structure that may be best described as a distorted trigonal bipyramid. The boron hydride [BEt3H]− and organolithium reagents, LiR, react with [Rh(allyltroppph)(metroppph)] in allylic alkylation reactions to yield anionic rhodate complexes [Rh(RCH2troppph)(metroppph)]− (R = H, Me, n-Bu, Ph) that formally have a d10 valence electron configuration. The rhodate [Rh(metroppph)2]− can be obtained directly by reduction of cation [Rh(metroppph)2]+ with alkali metals. In a sym-proportionation reaction [Rh(metroppph)2]+ and [Rh(metroppph)2]− give the neutral d9-[Rh(metroppph)2] radical (K = 1.1 × 107), which is not stable but decomposes with loss of H2 to give the allyl complex [Rh(allyltroppph)(metroppph)]. The structures in solution of [Rh(metroppph)2]+, [Rh(allyltroppph)(metroppph)], and [Rh(nBuCH2troppph)(metroppph)]− were determined by NMR techniques, which reveal that (i) they match the solid state structures and (ii) are rather similar to each other. This fact may explain the remarkable electronic flexibility of the rhodium centre, which changes reversibly its formal oxidation state from + I to 0 to − I at rather low negative potentials (ΔE01 = − 0.882 V; ΔE02 = − 1.298 V).
The chemistry of rhodium and iridium complexes in formally low oxidation states, that is 0 and − I, is comparatively little studied. In early works, strongly electron withdrawing ligands L like CO1 and fluorophosphanes2 were used for the synthesis of [ML4]− anions (M = Rh, Ir) with a formal d10 valence electron count on the metal. Only recently, the first stable rhodium and iridium d10-complexes with chelating phosphanes were prepared. 3a, 3b The synthesis of stable monomeric d9-[ML4] complexes was also only recently achieved. 3b, 4 These 17-electron complexes may have interesting properties as catalysts for the photolytic cleavage of water.5
We
have designed tropylidenylphosphanes (tropp)6 as a
new ligand system to allow the synthesis of stable rhodium4a
and iridium4b complexes in their formal oxidation states 0 and
−
I. These complexes were completely characterised including X-ray
analyses, which allowed us to study structural changes caused
by the addition or abstraction of single electrons (see also
ref. 3b). Furthermore, these [M(tropp)2]m complexes (M = Rh,
Ir; m
=
+ 1, 0, − 1) show very low redox potentials [M+1/M0: − 1.0
V (M = Rh), − 0.97 V (M = Ir); M0/M−1: − 1.2 V (M = Rh), − 1.4
V (M = Ir) ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) s. Ag/AgCl], which make them indeed potentially
interesting as redox catalysts.
s. Ag/AgCl], which make them indeed potentially
interesting as redox catalysts.
The vast structural data available for tetra-co-ordinated d8-rhodium(+I) complexes show these to be square planar, which is also supported by molecular orbital calculations.7 With the aim to provoke an even more anodic shift of the reduction potentials of rhodium tropp complexes, we wanted to prepare tetra-co-ordinated cationic rhodium(+I) compounds that are significantly distorted from their square planar configuration towards a tetrahedron.8 We expected that the ground state of the cation would be more destabilised than that of the product by such a distortion, leading to a lowering of the reduction potential. As will be seen later, this is not observed. However, we succeeded in the preparation of a strongly distorted tetra-co-ordinated rhodium complex and the chemistry of this compound and its derivatives is reported in this paper. In particular, the synthesis of a new substituted tropp ligand and a new method to prepare d10-rhodate complexes by nucleophilic attack on an allyl moiety co-ordinated to a rhodium(+I) centre are described.
Results and discussion
Synthesis of metroppph
The introduction of a methyl group into the olefinic unit of the central seven-membered ring of tropp, which co-ordinates to the metal centre, should increase the steric congestion and hence enforce a distorted co-ordination sphere. The synthesis of this new tropp ligand is outlined in Scheme 1. Starting from the readily available dibenzosuberenone 1, 10-bromo-5H-dibenzo[a,d]cyclohepten-5-one 2 was prepared by bromination of the double bond and subsequent HBr elimination.9 In the next step, the ketone functionality was protected by ketalisation using glycol in the presence of para-toluenesulfonic acid (pTosOH). In contrast to the literature procedure10 this conversion gave almost quantitatively compound 3. Reaction of 3 with excess Mg turnings in THF gave the corresponding Grignard reagent, which was reacted with an excess of methyl iodide. After addition of concentrated aqueous HCl and reflux for about 12 h, 10-methyl-5H-dibenzo[a,d]cyclohepten-5-one 4 was obtained in very good yield (90%) while the known procedure gave only 37% overall yield.10 The suberenone derivative 4 was reduced by modifying the literature procedure10 and then transformed into 5-chloro-10-methyldibenzo[a,d]cycloheptene 5. The addition of diphenylphosphine in toluene, followed by an aqueous work-up under basic conditions, led cleanly to the desired 5-diphenylphosphanyl-10-methyldibenzo[a,d]cycloheptene 6 as a racemic mixure of the R and S isomers in 86% isolated yield after recrystallisation from acetonitrile. Following our simplified nomenclature,4,6 we name this ligand metroppph where the superscript in front (me) indicates substituents bonded to the C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C bond of the seven-membered ring and the superscript at the end denotes the substituents bonded to phosphorus.
C bond of the seven-membered ring and the superscript at the end denotes the substituents bonded to phosphorus. | ||
| Scheme 1 Synthesis of metroppph. | ||
Synthesis of d8-rhodium metroppph complexes
The metroppph ligand rac-6 reacts quantitatively with the chloro-bridged binuclear rhodium cyclooctadiene (cod) complex 7 at room temperature to give an orange methylene chloride solution that contains a mixture of all possible diastereomers of the rhodium complexes 8, as indicated by the 31P NMR data given below (Scheme 2). The isomers cis-C2-8 and trans-C2-8 contain a C2 rotation axis, cis-meso-8 a mirror plane and trans-i-8 an inversion centre as symmetry elements and therefore each isomer should give rise to one 31P resonance signal split into a doublet by 103Rh–31P coupling. | ||
| Scheme 2 Synthesis of the cationic rhodium bis(metroppph) complex 9. | ||
Indeed, in the 31P NMR spectrum recorded at room temperature
only two exchange broadened doublets are seen,
which upon cooling to − 70 °C split into four doublets [1: δ31P
110.3, 1J(103Rh31P) = 212 Hz, 50%; 2: δ31P 110.0, 1J(103Rh31P) = 209
Hz, 35%; 3: δ31P 109.4, 1J(103Rh31P)≈207 Hz, 10%; 4: δ31P 109.5, 1J(103Rh31P)≈207 Hz, 50%]. In an 1H-NOESY NMR spectrum, it is seen
that eery isomer does exchange with every other component. Regarding
the different possible configurations of 8 in Scheme
2, apart from an intermolecular process ia
the solvated complex
[RhCl(metroppPh)S]
A (S = CH2Cl2), an intramolecular process
iaB can explain the pair-wise exchange between cis-C2-8
and trans-C2-8 on one hand and cis-meso-8 and trans-i-8
on the
other.11 All other exchanges must proceed ia intermolecular pathways
on which intermediates A or higher nuclear complexes
like C, which subsequently collapse to give all possible
stereoisomers, may appear. Note that when [Rh2(μ-Cl)2(cod)2]
7 is added, it also exchanges with all other components although
none of the
mixed dimer [Rh2(μ-Cl)2(troppph)(cod)] was directly observed. We did not assign the NMR signals to the different isomers nor determine the isomeric distribution. Crystals of 8 suitable for an X-ray analysis were not obtained. Note that in this context, the structure of [Rh2(μ-Cl)2(troppph)2] containing the unsubstituted troppph ligand was determined earlier and the major isomer showed the two phosphorus atoms in a cis arrangement.12
When 8 is dissolved in a co-ordinating solvent
like THF or MeCN, only one broadened 31P resonance is observed at 103.5 ppm [1J(103Rh31P) = 203 Hz].
Even when a large excess of rac-6 is reacted with 7 at elevated temperatures, no bis(metroppph) type complexes are formed, in contrast to reactions with the non-methyl-substituted ligand troppph, for which [RhCl(troppph)2] is obtained.4a, 12 However, when potassium hexafluorophosphate, KPF6, is added to an acetonitrile solution of 8, the dark red-violet bis(metroppph) complex 9 forms. Complex 9 is obtained in about 86% isolated yield; it is soluble in CH2Cl2 and acetonitrile but poorly soluble in THF. It can be crystallised by layering CH2Cl2 or CH3CN solutions with n-hexane. In solution, exclusively one isomer can be detected by NMR spectroscopy. The 103Rh–31P coupling constant [1J(103Rh31P) = 192 Hz is indicative of a cis isomer whereas the trans isomer [1J(103Rh31P) = 132 Hz] is predominant (80%) in solutions of the [Rh(troppph)2]+ cation.4
Interestingly, some of the protons of the annelated benzo groups and the phenyl substituents bonded to the phosphorus centres within each metroppph ligand show quite distinct 1H NMR signals in comparison to the uncomplexed ligand. For the ortho phenyl protons a (δ = 5.50) and b (δ = 7.65) (Fig. 1) in particular, this difference is significant and amounts to 2.15 ppm. It should be noted that this large difference is observed not only for the ortho protons but also for the entire phenyl rings. Indeed, for the latter, a remarkably different shielding is observed as a consequence of the conformation and arrangement of the phenyl substituents with respect to each other and to the benzo groups of the cycloheptatriene unit. A NMR determination of the three-dimensional solution structure reveals that one phenyl ring defines a plane with the Rh–P bond vector. This brings the ortho protons a into a deshielding region (indicated by x in Fig. 1) due to the magnetic anisotropy of the rhodium nucleus. The latter is caused by the dz2-type orbital at Rh(+I), which induces an unusually high frequency. On the other hand, the NOE between the PCH proton and the ortho protons b is remarkably weak, consistent with a dihedral angle between the corresponding phenyls of ca. ± 90° with respect to the Rh–P bond. In this conformational arrangement, the protons b are subject to a two-fold shielding anisotropy effect: (i) intraligand (y1) due to the neighbouring benzo group of the tropylidene unit and (ii) interligand (y2) due to a parallel arrangement of equivalent phenyl rings on each ligand in such a way that the protons of one ring lie in the shielding region of the other.
 | ||
| Fig. 1 Schematic representation of the cation of complex 9. a and b denote the set of different ortho-phenyl protons on each P centre. For a detailed picture of the structure see Fig. 2. | ||
We have no evidence for the formation of penta co-ordinated
[Rh(tropp)2L]+ complexes (L = MeCN or THF) even
though such species are formed with sterically less encumbered
tropp-type ligands.4c Indeed, the exclusive formation of
the chiral (C2 symmetry) R,R and S,S isomers, respectively,
was established by an X-ray analysis (ide infra). The meso isomer
is not observed, probably because of even higher steric congestion, which prevents its formation. In the 103Rh NMR spectrum
of a CH2Cl2 solution of 9, one sharp resonance signal
(triplet) at + 162 ppm is observed, which is typical for
16-electron rhodium tropp complexes.
Structure of [Rh(metroppph)2]PF6·2CH3CN (9)
The salt [Rh(metroppph)2]PF69 crystallises with two crystallographically independent pairs of enantiomers in the unit cell. All bonds and angles are equal within the limits of experimental accuracy and we discuss therefore only one of the two cations. Additionally, two acetonitrile molecules are included, which have no close contact with the rhodium centre. Further crystallographic details are given in Table 1, selected bond lengths and angles are listed in Table 2. In Fig. 2, an ORTEP 3 plot13 of the cation of 9 is shown.| 9 | 10 | |
|---|---|---|
| Chemical formula | C58H49F6NP3Rh | C55.5H45.40Cl1.50P2Rh |
| Formula weight | 1069.80 | 942.45 |
| Crystal system | Triclinic | Orthorhombic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | C2/c |
| a/Å | 10.844(2) | 45.90(5) |
| b/Å | 20.697(4) | 10.159(10) |
| c/Å | 22.722(5) | 19.773(19) |
| α/° | 91.629(13) | 90 |
| β/° | 91.462(13) | 102.27(8) |
| γ/° | 101.890(12) | 90 |
| U/Å3 | 4985.7(17) | 9009(16) |
| Z | 4 | 8 |
| T/K | 293(2) | 293(2) |
| μ/mm−1 | 0.501 | 4.842 |
| Reflections collected | 28990 | 4804 |
| Independent reflections | 14653 [R(int) = 0.0378] | 4620 [R(int) = 0.0428] |
| Final R indices [I>2σ(I)] | ||
| R1 | 0.036 | 0.046 |
| wR2 | 0.072 | 0.113 |
| Rh1–P1 | 2.226(1) | Rh1b–P1c | 2.212(1) |
| Rh1–P1a | 2.260(1) | Rh1b–P1b | 2.266(2) |
| Rh1–C5 | 2.337(4) | Rh1b–C5c | 2.341(4) |
| Rh1–C4 | 2.362(4) | Rh1b–C4c | 2.381(4) |
| Rh1–C5a | 2.404(4) | Rh1b–C5b | 2.401(4) |
| Rh1–C4a | 2.451(4) | Rh1b–C4b | 2.508(4) |
| P1–C17 | 1.818(5) | P1b–C23b | 1.812(5) |
| P1–C23 | 1.819(4) | P1b–C17b | 1.839(4) |
| P1–C1 | 1.878(4) | P1b–C1b | 1.874(4) |
| C4–C5 | 1.372(6) | C4b–C5b | 1.373(6) |
| P1a–C23a | 1.825(5) | P1c–C23c | 1.817(4) |
| P1a–C17a | 1.830(4) | P1c–C17c | 1.819(4) |
| P1a–C1a | 1.858(4) | P1c–C1c | 1.872(4) |
| C4a–C5a | 1.380(6) | C4c–C5c | 1.375(6) |
| P1–Rh1–P1a | 94.18(4) | P1c–Rh1b–P1b | 93.44(4) |
| P1–Rh1–C5 | 90.2(1) | P1c–Rh1b–C5c | 90.8(1) |
| P1A–Rh1–C5 | 166.7(1) | P1b–Rh1b–C5c | 169.8(1) |
| P1–Rh1–C4 | 90.1(1) | P1c–Rh1b–C4c | 89.7(1) |
| P1a–Rh1–C4 | 133.3(1) | P1b–Rh1b–C4c | 136.8(1) |
| C5–Rh1–C4 | 34.0(1) | C5c–Rh1b–C4c | 33.8(1) |
| P1–Rh1–C5a | 167.0(1) | P1c–Rh1b–C5b | 164.1(1) |
| P1a–Rh1–C5a | 89.7(1) | P1b–Rh1b–C5b | 89.2(1) |
| C5–Rh1–C5a | 88.8(1) | C5c–Rh1b–C5b | 89.3(1) |
| C4–Rh1–C5a | 96.2(1) | C4c–Rh1b–C5b | 99.1(1) |
| P1–Rh1–C4a | 134.5(1) | P1c–Rh1b–C4b | 131.8(1) |
| P1a–Rh1–C4a | 89.3(1) | P1b–Rh1b–C4b | 89.8(1) |
| C5–Rh1–C4a | 96.6(2) | C5c–Rh1b–C4b | 94.3(2) |
| C4–Rh1–C4a | 119.3(2) | C4c–Rh1b–C4b | 119.1(1) |
| C5a–Rh1–C4a | 33.0(1) | C5b–Rh1b–C4b | 32.4(1) |
| C17–P1–C23 | 102.6(2) | C23b–P1b–C17b | 104.2(2) |
| C17–P1–C1 | 102.8(2) | C23b–P1b–C1b | 99.6(2) |
| C23–P1–C1 | 101.6(2) | C17b–P1b–C1b | 103.3(2) |
| C17–P1–Rh1 | 125.0(1) | C23b–P1b–Rh1b | 110.7(1) |
| C23–P1–Rh1 | 111.8(1) | C17b–P1b–Rh1b | 125.8(1) |
| C1–P1–Rh1 | 110.3(1) | C1b–P1b–Rh1b | 110.1(1) |
| C23a–P1a–C17a | 105.4(2) | C23c–P1c–C17c | 106.9(2) |
| C23a–P1a–C1a | 101.8(2) | C23c–P1c–C1c | 100.7(2) |
| C17a–P1a–C1a | 102.6(2) | C17c–P1c–C1c | 103.3(2) |
| C23a–P1a–Rh1 | 110.2(1) | C23c–P1c–Rh1b | 110.7(1) |
| C17a–P1a–Rh1 | 124.6(1) | C17c–P1c–Rh1b | 122.6(1) |
| C1a–P1a–Rh1 | 109.8(1) | C1c–P1c–Rh1b | 110.3(1) |
 | ||
| Fig. 2 Molecular structure of one of the crystallographically independent cations of 9. Hydrogen atoms are omitted for clarity. Selected bond lengths and angles are given in Table 2. | ||
The two phosphorus centres occupy the cis positions if an
idealised square planar conformation is taken into consideration.
However, the co-ordination sphere of the rhodium centre
is strongly distorted towards a tetrahedron. The angle φ
between the two planes that run through each phosphorus centre,
the rhodium centre and the midpoint of each of the metal-bound
CC bonds amounts to 42°. Hence, the co-ordination
is halfway between a square plane (φ
= 0°) and a tetrahedron (φ
= 90°) and is to our knowledge the second strongest deviation
observed to date for tetra-co-ordinated d8-rhodium complexes.8
While the Rh–P distances [mean 2.241(1) Å; max. difference
Δ: 0.054 Å] lie within the usual range (≈2.30 Å),14
the steric congestion
is more clearly seen in the unusually long Rh–C distances
[mean: Rh–C4: 2.426(4) Å, Δ
= 0.066 Å; Rh–C5: 2.371(4) Å, Δ
= 0.146 Å],
which are about 0.24 Å longer than the usually observed bond lengths
(≈2.15 Å).14 The variation of the individual Rh–P or Rh–C4/C5
bonds expressed by the difference Δ between the shortest
and longest bond is also considerable.14
The co-ordinated CC bonds
[mean: 1.375(6) Å] are not very elongated when compared to
the free ligand.6 The sum of bond angles around the carbon centre
C4 shows only a weak pyramidalisation (by less than 4°), indicating only a weak d → p(π) back donation from the metal centre
to the CC unit.
Synthesis of the allyl complex [Rh(allyltroppph)(metroppph)] (10)
When potassium tert-butoxide is added to a solution of R,R-9 (S,S-9) in THF (Scheme 3) an immediate colour change from deep red to intense orange occurs and 10, 10′ are formed quantitatively. | ||
| Scheme 3 Synthesis of the allyl complex 10 and allylic alkylation giving 12a–d. | ||
In the reaction solution, only two products are observed by 31P NMR in a ca. 28:1 ratio, showing each signal as a doublet of doublets [major: δ 31P 88.0, 1J(103Rh31P) = 172.0 Hz, 79.0 1J(103Rh31P) = 159 Hz; 91.7 1J(103Rh31P) = 168 Hz, 81.4 1J(103Rh31P) = 158 Hz]. The very similar chemical shifts and coupling constants observed suggest the formation of two structurally related isomers.
Recrystallisation from CH2Cl2–n-hexane gave complex 10 in almost quantitative yield. The structure was determined by an X-ray analysis (Fig. 4) and we reasonably assume that this structure corresponds to the major isomer 10 (see Scheme 3) observed in solution. However, when pure re-crystallised 10 is dissolved in CH2Cl2, both isomers are again found in the same ratio (28:1). Because of the similarity of the 31P NMR data, we assign to the minor component 10′ a structure in which the intact metroppph ligand has rotated by 180° when compared to the major product. Consequently, the methyl group points towards the CH2 terminus of the allyl moiety as shown in Scheme 3. For this isomerisation, an interesting possibility would have consisted of an intra-molecular deprotonation of the methyl substituent of one tropp ligand by the allyl group of the other. However, 2D-NOESY spectra show no cross-peaks for the corresponding protons and hence give no indication for such a phenomenon.
Most informative are the heteronuclear correlations. For the 103Rh–1H HMQC (heteronuclear multiple quantum coherence) NMR data of rhodium metroppph complexes, the three expected 103Rh–1H cross-peaks per inequivalent ligand are found for the protons of the PCH and the olefinic H and Me group. However, there is an additional cross-peak for 10, indicating the formation of a CH2 unit with diastereotopic protons rather than an additional Me group. The low frequency 103Rh shift (δ = −370) is consistent with the formulation of an η3-allyl moiety. This is further confirmed by the 13C, 1H one- and multiple-bond correlations showing a tertiary C5H, quaternary C4 and secondary C16H2 with δ = 61.7, 103.0 and 58.1, respectively (see Fig. 3). Such values are typically encountered for η3-allyl groups.
 | ||
| Fig. 3 Schematic representation of 10 illustrating two- and multiple-bond correlations. | ||
After having identified some structural features of the complex, we now turn to an analysis of the solution structure. As such, the two-bond coupling constants between ligand atoms are the most valuable tool in the determination of the configuration at the rhodium centre. In this respect, 2J(31P31P) = 29 Hz indicates a cis arrangement of the P1 and P1a centres. Within the constraints of an assumed trigonal bipyramid, this means that either both phosphorus are equatorial or one is equatorial and the other one axial. Placing the allyltropp ligand with its P1 centre in an axial position brings both allyl termini C5 and C16 in equatorial positions. Hence, the angle between these two carbon atoms and the P1a nucleus of the metropp would have similar values. However, this is in contrast to the observed coupling constants 2J(31P13C) of 41.8 and 11.0 Hz for the CH and CH2 carbons, respectively. Indeed, the former value indicates a transoid arrangement. Accordingly, the allyl C5H carbon is most probably located in an axial position trans to the P1a centre of the metropp ligand. Consequently, the allyl C16H2 and the olefinic bond are equatorial. A retention of the chirality of the ligands requires that the angle C5a–Rh–P1 be larger than C4a–Rh–P1. This is indeed reflected by the larger coupling constant values [2J(31P13C) = 21.8 Hz] for the C5aH carbon relative to the C4aMe [2J(31P13C) = 10.3 Hz].
The NOE results are fully consistent with these results, the olefinic H at C5a is close to both anti protons Ha of the allyl moiety, whereas the syn proton Hs shows a short separation to the ortho proton of the in-plane phenyl group attached to the intact metroppph ligand. On the basis of the NMR data, with the exception of phenyl ring flips (rather than rotations), the solution structure can be assumed as being quite static, showing no indication of the dynamic behaviour often encountered in allyl and penta co-ordinated organometallic chemistry.
The allyl complexes 10,10′ do not react with water or alcohol and resists treatment with diluted acetic acid. However, when stronger acids like HO2CCF3, aqueous HPF6, or HBF4 are added, a clean re-protonation occurs and the cationic complex 9 is reformed quantitatively.
Structure of [Rh(allyltroppph)(metroppph)] (10)
An ORTEP 3 plot of the structure of 10 is shown in Fig. 4. Crystallographic details are given in Table 1, selected bond lengths and angles are listed in Table 3. The rhodium centre lies within a highly distorted co-ordination sphere, which we describe here as trigonal pyramidal where P1a, C5 occupy the axial positions (P1a–Rh1–C5 159.8°) and the olefinic unit C4aC5a,
P1, and C16 the equatorial positions. | ||
| Fig. 4 Molecular structure of 10. Hydrogen atoms are omitted for clarity. Selected bond lengths and angles are given in Table 3. | ||
| Rh1–C4 | 2.168(4) | P1–C23 | 1.839(5) |
| Rh1–C5a | 2.203(5) | P1–C1 | 1.889(4) |
| Rh1–C5 | 2.208(4) | P1a–C17a | 1.839(4) |
| Rh1–P1a | 2.278(2) | P1a–C23a | 1.843(5) |
| Rh1–C16 | 2.293(6) | P1a–C1a | 1.879(4) |
| Rh1–C4a | 2.295(4) | C4–C5 | 1.437(6) |
| Rh1–P1 | 2.341(2) | C4a–C5a | 1.424(6) |
| P1–C17 | 1.823(5) | ||
| C4–Rh1–C5a | 109.1(2) | C5–Rh1–P1 | 88.9(1) |
| C4–Rh1–C5 | 38.3(2) | P1a–Rh1–P1 | 101.0(1) |
| C5a–Rh1–C5 | 90.6(2) | C16–Rh1–P1 | 122.0(1) |
| C4–Rh1–P1a | 123.2(1) | C4a–Rh1–P1 | 111.6(1) |
| C5a–Rh1–P1a | 90.4(1) | C17–P1–C23 | 102.0(2) |
| C5–Rh1–P1a | 159.8(1) | C17–P1–C1 | 99.4(2) |
| C4–Rh1–C16 | 37.0(2) | C23–P1–C1 | 100.3(2) |
| C5a–Rh1–C16 | 86.5(2) | C17–P1–Rh1 | 121.0(2) |
| C5–Rh1–C16 | 64.1(2) | C23–P1–Rh1 | 122.7(1) |
| P1a–Rh1–C16 | 95.9(1) | C1–P1–Rh1 | 107.3(1) |
| C4–Rh1–C4a | 135.0(2) | C17a–P1a–C23a | 100.2(2) |
| C5a–Rh1–C4a | 36.8(2) | C17a–P1a–C1a | 101.0(2) |
| C5–Rh1–C4a | 100.9(2) | C23a–P1a–C1a | 103.8(2) |
| P1a–Rh1–C4a | 91.9(1) | C17a–P1a–Rh1 | 121.9(1) |
| C16–Rh1–C4a | 122.8(2) | C23a–P1a–Rh1 | 119.1(2) |
| C4–Rh1–P1 | 90.1(1) | C1a–P1a–Rh1 | 108.2(2) |
| C5a–Rh1–P1 | 147.4(1) |
The intersection of the plane defined by P1a, Rh1 and the
midpoint of the C4aC5a unit and the P1–Rh1–C4 plane, gives an angle φ of 78°. The phosphorus rhodium distances [Rh–P1 2.341(2);
Rh–P1a 2.278(2) Å] fall within the expected range, while the
olefinic [Rh1–C5a 2.203(5); Rh1–C4a 2.295(4) Å] and the allylic
[Rh1–C5 2.208(4); Rh1–C4 2.164(4); Rh1–C16 2.293(6) Å] bond
lengths are longer than usually observed. Both the CC and
the allyl group are unsymmetrically bound to the rhodium centre
(ΔRh–C≈0.09 Å). However, the almost equal C–C bond lengths [C4–C5
1.437(6); C4–C16 1.421(6) Å] within the allyl moiety, when compared
to the corresponding distances in the intact metroppph
ligand [C4a–C5a 1.424(6); C4a–C16a 1.513(7) Å], clearly indicate
the η3 binding
mode. An alternative possibility would have been a σ(η1),π(η2)-enyl mode in which the CH2
group is σ-bonded and the CC unit π-bonded to the rhodium
centre
(see, however, ref. 15b). Also, the sum of bond angles at C4 (360°)
when compared to C4a (353.3°) is in line with the η3-description
of the allyl group.
In summary, it can be concluded that the solution structure
of 10 and its solid state structure (ide infra) are very closely related.
Synthesis of d10-rhodate complexes
The nucleophilic attack on co-ordinated allyl groups has been widely studied, in particular for cationic 16-electron palladium complexes, [Pd(C3R5)L2]+.16 Little is known about the corresponding reaction of isoelectronic but neutral rhodium complexes. In recent work, it was shown that rhodium complexes of the Wilkinson type prepared in situ from [RhCl(PPh3)3] and P(OR)3 may have an interesting synthetic potential as catalysts for allylic alkylation reactions.15 However, it was proposed that π(η3)-allyl intermediates are not the reactive species but rather a σ(η1),π(η2)-enyl complex with a formal oxidation state of + III on the rhodium centre.15bIn this context, the reaction of the rhodium(+I) complex 10 with various nucleophiles LiR 11a–d was particularly interesting (Scheme 3). Note that 10 is within formal concepts an electronically saturated 18-electron metal complex in which the η3-coordination mode of the allyl moiety is clearly established. In view of the vast literature available on the nucleophilic attack on 16-electron complexes, which are mostly cationic, this reaction seemed not that evident with the neutral 18-electron complex 10. However, in all cases the nucleophile attacks the CH2 terminus of the co-ordinated allyl moiety and the d10-rhodate complexes 12a–d are formed. The syntheses of 12a and 12c proceeded even at room temperature within less than 5 min. The two complexes were isolated as highly oxygen- and water-sensitive deep burgundy-red micro-crystalline substances. The formation of the compounds 12b and 12d is slow and these could only be characterised by NMR spectroscopy in solution. Other nucleophiles like amides, alcoholates, or thiolates did not react. During these reactions, the formal valence electron count does not change—all complexes are 18-electron species—and the formal oxidation state of the rhodium centre is diminished from + I to − I. Consequently, the rhodate complex 12a could also be prepared from the cationic d8-rhodium complex 9 by two-electron reduction with elemental lithium, sodium or potassium in THF. Unfortunately, we did not succeed in growing crystals suitable for an X-ray analysis of one of these compounds.
All complexes show fairly complex NMR spectra and as an example, complex 12c was carefully investigated by various techniques. For this molecule the solution structure can be determined unambiguously as there is not a single atom related to any other by symmetry. A complete assignment of the 103Rh, 31P, 13C, and 1H resonances was possible on the basis of 103Rh–1H, 31P–1H and 13C–1H one- and multiple-bond correlations in combination with 1H–1H TOCSY and NOESY measurements. Subtle effects, such as chirality of the ligand, phenyl ring orientations and pentyl side chain conformations could all be determined. With respect to the former two points, it turns out that: (i) both ligands in 12c have the same absolute configuration and (ii) the phenyl ring orientations are as already discussed for 9. The interligand relationship is interesting because it determines the structure around the central rhodium atom. Starting from a tetrahedral situation (see A in Fig. 5), one expects to have the olefinic protons close to each other and to the ortho protons of the adjacent arene ring of the bis(benzo)cycloheptatriene. However, this is clearly not the case as can be seen from Fig. 6. In fact, the distances a1 between the olefinic proton on the pentyltroppph ligand group and the methyl group and from the latter to the olefinic proton of the metroppph ligand, a2, seem to be quite similar. A similar short triangulation, b1, b2, to the olefinic proton and one arene proton of the opposing metroppph ligand is seen for one of the diastereotopic methylene H of the CH2 pentyl group directly linked to the central seven-membered ring. This indicates a situation (B) where the tetrahedron is strongly distorted towards a square planar arrangement. Indeed, the structure found for the Rh(+I) compound 9 fits exceptionally well with the constraints of the NOE data.
 | ||
| Fig. 5 Schematic representation of two possible structures of 12c in solution. (A) tetrahedral structure; (B) distorted planar structure resembling closely the one of 9. The HH distances a1, a2, b1, and b2 are indicated by dotted lines. | ||
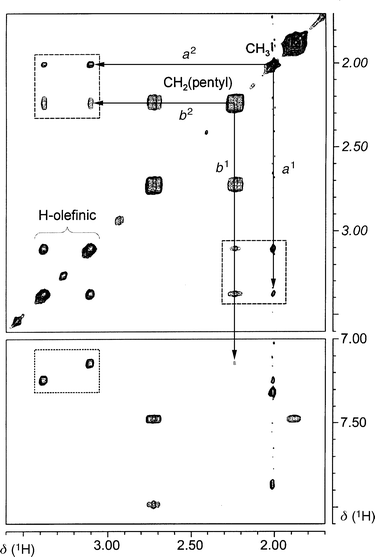 | ||
| Fig. 6 Contour plots of sections from the 2D NOESY spectrum (400.13 MHz, τmix = 600 ms) recorded for 12c. The dotted box in the lower part shows two cross-peaks due to intra-ligand NOEs between the two olefinic protons and adjacent ortho-protons of the benzoheptene moiety. Note that cross-peaks in the opposite corners of the box stemming from the inter-ligand NOEs expected for a regular tetrahedral structure are absent. The dashed boxes in the upper part of the spectrum show cross-peaks of similar intensity from each of the two olefinic protons to the methyl and to one of the diastereotopic methylene protons of the pentyl group. These observations are in agreement with the distorted structure described in the text. | ||
In the 103Rh spectra of 12a, a triplet centered at δ = − 344 is observed. Complex 12c shows a doublet of doublets at δ = −325. Both chemical shifts fall within the same range as the one found for the allyl complex 10 while 16-electron rhodium tropp complexes like 9 show resonances shifted about 500 ppm to higher frequencies.
Electrochemical investigations and observation of the d9-rhodium tropp complex
In previous work, we have shown that the troppph ligand stabilises rhodium and iridium complexes in their formally low oxidation states.4 In particular, the redox potentials M+1/M0 and M0/M−1 are considerably lowered (up to 1 V) when compared to reported systems.17 We expected that the highly tetrahedrally distorted cation d8-[Rh(metroppph)2]+ in 9 would be reduced to the corresponding d9 and d10 valence states at even lower potentials (ide supra). The cyclic voltammogram
of 9 in THF solution (1 × 10−3 M) containing 0.25 mol n-Bu4NPF6
as electrolyte at room temperature is shown in Fig. 7. Using
a scan rate of 100 mV s−1,
two almost reversible redox waves
at − 0.882 V (ΔEred/ox1
= 82 mV) and − 1.298 V (ΔEred/ox2
= 85 mV) are
recorded (potentials s. Ag/AgCl; reference: ferrocene/ferrocenium). The first wave corresponds to the redox couple [Rh(metroppph)2]+(9)/[Rh(metroppph)2] (13), the second to the couple
13/[Rh(metroppph)2]−(12a) (Scheme
4). | ||
| Fig. 7 Cyclic
voltammograms of 9 measured in THF (20 °C); scan rate: 100 mV s−1; E s. Ag/AgCl. | ||
![Disproportionation and decomposition of d9-[Rh(metroppph)2]
13.](/image/article/2001/NJ/b002906h/b002906h-s4.gif) | ||
| Scheme 4 Disproportionation and decomposition of d9-[Rh(metroppph)2] 13. | ||
From the ΔE of the two redox processes, the formation constant
Kf of the reaction 9
+
12a![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) 2 13 was calculated to be 1.1 × 107
at 298 K. Indeed, this reaction is the most convenient
way to prepare 13 (see Scheme 4). However, the rhodium(0)
complex 13 is not stable but decomposes cleanly—formally
with loss of H2—at room temperature in about one week, in
15 min at 70 °C, by a yet unclarified pathway to the allyl complex
10.
2 13 was calculated to be 1.1 × 107
at 298 K. Indeed, this reaction is the most convenient
way to prepare 13 (see Scheme 4). However, the rhodium(0)
complex 13 is not stable but decomposes cleanly—formally
with loss of H2—at room temperature in about one week, in
15 min at 70 °C, by a yet unclarified pathway to the allyl complex
10.
Despite the strong distortion of the cation [Rh(metroppph)2]+ in 9, the first reduction to the neutral complex 13 shifts only about 0.12 V to less negative potentials when compared to that of the sterically less encumbered cation [Rh(troppph)2]+, which has a square planar co-ordination sphere.4a The second reduction appears at about the same potential as that of this latter complex (only a slight cathodic shift of about − 0.08 V is observed). These experiments clearly show that a distortion of about 40° is not sufficient to cause a significant anodic shift of the reduction potential in this type of rhodium complexes. Furthermore, the electron-donating effect of the methyl group18 may partly counterbalance the diminution of the first redox potential induced by the geometric distortion.
Conclusions
A high yield synthesis of a sterically demanding new tropylidene phosphane, metroppph, was developed. This ligand allowed the preparation of the tetra-co-ordinated cationic rhodium complex 9, which is obtained as a racemic mixture of the chiral R,R and S,S enantiomers exclusively. The cation 9 shows a deep red-violet colour in contrast to the bright red-orange colour usually observed for tetra-co-ordinated cationic d8-rhodium complexes. The co-ordination sphere deviates strongly from planarity with φ = 42° being almost precisely between a square plane (φ = 0°) and a tetrahedron (φ = 90°). Comparable distorted structures were also determined for the neutral d9-[M(troppph)2] complexes (M = Rh, Ir) and anionic d10-[M(troppph)2]− compounds, the parent cationic complex d8-[M(troppph)2]+ being planar.4Unfortunately, the expected shift to less negative potentials for the reduction of the d8-rhodium(+I) to d9-rhodium(0) state is not observed. The latter complex, d9-[Rh(metroppph)2] 13, characterised by its deep green colour, is less stable compared to the sterically less encumbered complex d9-[Rh(troppph)2] and decomposes with formal loss of H2 to the allyl complex [Rh(allyltroppph)(metroppph)] 10. This allyl complex may be prepared more conveniently in almost quantitative yield from cation 9 and KOBut. Despite the electronic saturation of 10 (18 valence electrons), nucleophilic attack by lithium organyls, LiR, cleanly afforded new anionic rhodate complexes d10-[Rh(RCH2troppph)(metroppph)]−12a–c in which the R group has been incorporated into the former methyl group of one metroppph ligand.
The structures of 9, 10 and 12c were determined in solution by NMR techniques. All complexes are remarkably rigid; even the phenyl groups seem to be locked into specific conformations. The solid state structures of cation 9 and of the allyl complex 10 are fully confirmed in solution as well. Furthermore, it could be demonstrated that the anionic complex 12c must have a structure in solution very close to the one of cation 9, that is a structure deviating considerably from the tetrahedral one that is expected for a tetra-co-ordinated d10-metal centre. In summary, there is only relatively little structural change between 9, 10 and 12. This may explain some of their properties, such as low redox potentials or facile nucleophilic attack on the allyl moiety, reactions by which the formal oxidation state of the rhodium centre is changed. Obviously the steric constraints imposed by the tropp-type ligands make the complexes sterically rigid but the metal centres electronically flexible.
Experimental
General methods
NMR spectra were taken on a Bruker DPX Avance Series 250–400 MHz. 1H- and 13C-chemical shifts are calibrated against the solvent signal (CDCl3: 1H-NMR: 7.27 ppm; 13C-NMR: 77.23 ppm; CD2Cl2: 1H-NMR: 5.32 ppm; 13C-NMR: 54.00 ppm; THF-d8: 1H-NMR: 3.58 ppm; 13C-NMR: 67.57 ppm). 31P chemical shifts are calibrated against 85% H3PO4 as external standard. 103Rh-NMR spectra are referenced to Ξ = 3.16 MHz, where TMS appears at exactly 100.000000 MHz. 103Rh–1H, 13C–1H correlations were measured using standard HMQC and HMBC (heteronuclear multiple bond coherence) methods, 31P–1H 2D spectra using the COSY method with 1H detection, 1H–1H TOCSY with MLEV 17 mixing and the 1H–1H NOESY with a mixing time of 600 ms. All NMR spectra were measured at ambient temperature. Cyclic voltammograms were acquired with an EG&G potentiostat model 362. UV/Vis spectra were recorded on a Perkin Elmer UV/Vis/NIR-spectrometer Lambda 19 and mass spectra on a Finnigan MAT SSQ 7000 (EI: 70 eV). All chemicals were used as purchased from Aldrich or Fluka, solvents were purified and dried following standard procedures. 10-Bromo-5H-dibenzo[a,d]cyclohepten-5-one 2 was prepared following a literature procedure.9Crystallographic analysis of 9 and 10
The data sets for the single-crystal X-ray studies were collected on a Stoe Image Plate System (9) or a fully automated Siemens-Stoe AED2 four-circle diffractometer (10). All calculations were performed using the SHELXS-86 and SHELXS-93 program suites. The specific data for the crystals and refinements are collected in Table 1.CCDC reference number 440/204. See http://www.rsc.org/suppdata/nj/b0/b002906h/ for crystallographic files in .cif format.
Synthesis of compounds
CH), 7.44–7.28
(m, 5 H, CHar), 4.27–4.15 (m, 2 H, CH2exo), 3.83–3.70 (m,
2 H, CH2endo). 13C-NMR (CDCl3, 75.4 MHz): δ
= 139.3 (Cqar), 138.9
(Cqar), 133.7 (CHar), 132.7 (Cqar), 132.3 (Cqar), 130.3
(CHar), 129.4 (CHar), 128.8 (CHar), 128.2 (CHar), 127.9 (CHar),
127.7 (CHar), 124.7 (CBr), 123.8 (CHar), 106.0 (CH), 65.0 (CH2endo), 64.4 (CH2exo).acuo to yield a slightly brownish oil, which
crystallised upon layering with a small amount of diethyl
ether (5, 4.41 g, 91% from 4). Compound 5 is obtained as a
mixture of exo and endo isomers. Mp: 72 °C. MS m/z: 242 (M+,
5%), 240 (M+, 8%), 205 (10-methyl-5H-dibenzo[a,d]tropylium+,100%).
1H-NMR (CDCl3, 300 MHz): δ
= 6.24 (s, 1 H, CHClmaj), 5.60
(s, 1 H, CHClmin), 2.59 (s, 1 H, CH3maj), 2.53 (s, 1 H, CH3min).
13C-NMR (CDCl3, 75.4 MHz): δ
= 138.1, 137.2, 137.1,
136.6, 134.9, 132.8, 131.5, 129.9, 129.5,
129.0, 128.8, 128.6, 128.55, 128.4, 128.35, 128.3, 128.2, 127.8, 127.6, 127.55, 127.5, 127.0,
126.7, 126.4, 125.3 (CHmin), 125.2 (CHmaj), 123.3, 123.0, 67.4
(CHClmaj), 61.1 (CHClmin), 25.0 (CH3maj), 24.3(CH3min).ia syringe
into a solution of 5 (2.4 g, 10 mmol) in dry toluene (100
ml). A white precipitate formed immediately. After 2 h of stirring, a carefully degassed 5% aqueous solution of sodium carbonate was added, the organic layer separated, dried over sodium sulfate and the solvent removed under reduced pressure leaving crude 6. After recrystallisation from acetonitrile, 6 was obtained as colourless crystals (3.35 g, 86%). Mp: 127 °C. MS m/z: 390 (M+, 42%), 205 (10-methyl-5H-dibenzo[a,d]tropylium+, 100%). 31P-NMR (CDCl3, 121.5 MHz): δ
=
− 11.7. 1H-NMR (CDCl3, 300 MHz): δ
= 7.48–7.42 (m, 1 H, CHar), 7.34–6.94 (m, 17 H, CHar), 7.14 (s, 1 H, CH), 4.80 [d, 2J(PH) = 6.2 Hz, CHPR2], 2.51 (s, 3 H, CH3). 13C-NMR (CDCl3, 75.4 MHz): δ
= 139.8 [d, J(CP) = 8.2 Hz, Cqar], 138.9 [d, J(CP) = 8.8 Hz, Cqar], 138.5 [d,
J(CP) = 20.1 Hz, Cqar]), 138.1 [d, J(CP) = 19.5 Hz, Cqar], 138.0
[d, J(CP) = 5.2 Hz, Cqar], 137.1 [d, J(CP) = 4.6 Hz, Cqar], 133.7 [d, J(CP) = 20.4 Hz, CHar], 133.4 [d, J(CP) = 19.6 Hz, CHar], 130.4 [d, J(CP) 5.8 Hz, CHar], 129.7 [d, J(CP) = 3.4 Hz, CHar], 129.2 [d, J(CP) = 3.0 Hz, CHar], 129.0 [d, J(CP) = 1.8 Hz, CHar],
128.3 (s, CHar), 128.2 (s, CHar), 127.8 [d, J(CP) = 2.1 Hz, CHar],
127.8 [d, J(CP) = 2.1 Hz, CHar], 127.7 [d, J(CP) = 1.8 Hz, CHar], 127.3 (s, CH), 126.8 [d, J(CP) = 1.0 Hz, CHar], 126.3 [d, J(CP) = 1.3 Hz, CHar], 126.4 (s, CBr), 126.2 [d, J(CP) = 1.5 Hz,
CHar], 56.9 [d, 1J(PC) = 20.1 Hz, CHPR2], 25.0 (s, CH3).| 103Rh | 31P | CHCR | CHCR | CHCR | CHPPh2 | Mp/ °C | λmax/nm | |
|---|---|---|---|---|---|---|---|---|
| 6 | − 11.7 (s) | 127.3 (s) | 126.4 (s) | 25.0 (s) | 56.9 [d, 1J(PC) = 20.1] | 127 | ||
| 7.14 (s) | 2.51 (s) | 4.80 [d, 2J(PH) = 6.2] | ||||||
| 9 | 162 (t) | 84.5 [d, 1J(RhP) = 192] | 99.9 (m) | 123.7 (m) | 30.0 (s) | 58.8 (m) | 247–252 | 510 |
| − 142.7 [h, 1J(PF) = 708] | 6.73 (s) | 2.75 (s) | 4.85 (m) | (dec.) | 324 | |||
| 10 (maj) | −370 (dd) | 88.0 [dd, 1J(RhP1) = 172, | 61.7 [dd, 1J(RhC) = 7.5, | 103.0 (m) | 58.1 (m) | 54.2 [d, 1J(PC) = 14.5] | 265–268 | 329 |
| 2J(PP) = 29.0] | 3J(P2C) = 1.3] | 2.97 (m, CH exo) | 5.06 [d, 2J(PH) = 12.0] | (dec.) | ||||
| 2.68 [dd, 1J(PH) = 8.7, | 2.73 (m, CH endo) | |||||||
| 2J(RhH) = 1.4] | ||||||||
| 79.0 [dd, 1J(RhP2) = 159, | 79.5 [dd, 3J(P1C) = 22.0, | 64.3 [ddd, 2J(P2C) = | 29.8 [d, 2J(RhC) = 4.5] | 55.7 [d, 1J(PC) = 15.4] | ||||
| 2J(PP) = 29.0] | 1J(RhC) = 7.0] | 41.7, 1J(RhC) = 10.2, | 1.99 (s) | 4.98 [d, 2J(PH) = 12.8] | ||||
| 3.92 (m) | 2J(P1C) = 6.5] | |||||||
| 10′ (min) | 91.7 [dd, 1J(RhP1) = 168, | |||||||
| 2J(PP) = 26.1] | ||||||||
| 81.4 [dd, 1J(RhP2) = 158, | ||||||||
| 2J(PP) = 26.1] | ||||||||
| 12a | −344 (t) | 87.4 [d, 1J(RhP) = 180 | 70.6 [ddd, 2J(PalterC) = 42.2, | 48.5 [d, 1J(RhC) = 12.6] | 30.8 (m) | 57.6 [dd, 1J(PC) = 10.3, | 502 | |
| 1J(RhC) = 9.1, 2J(PipsoC) = 1.8] | 1.86 (s) | 2J(RhP) = 4.3] | 377 | |||||
| 3.13 [d, 2J(PH) = 4.3] | 4.44 [d, 2J(PH) = 9.3] | 292 | ||||||
| 12b | 85.8 [dd, 1J(RhP1) = 184, | |||||||
| 2J(PP) = 7.8] | ||||||||
| 85.2 [dd, 1J(RhP2) = 183, | ||||||||
| 2J(PP) = 7.8] | ||||||||
| 12c | −325 (dd) | 86.2 [dd, 1J(RhP1) = 182, | 68.4 (m) | 54.4 [d, 1J(RhC) = 12.5] | 42.8 (m, Cα), 32.6 (s, Cγ), 30.5 | 57.6 [dd, 1J(PC) = | 497 | |
| 2J(PP) = 7.5] | 3.49 (m) | (m, Cβ), 23.9 (Cδ), 14.9 (s, Cε) | 10.5, 2J(RhP) = 4.1] | 372 | ||||
| 2.53 (m, CαH), 2.06 (m, CαH), | 4.38 [d, 2J(PH) = 10.2] | 284 | ||||||
| 1.65 (m, CβH), 1.39–1.12 (m, | ||||||||
| 5H, CβH, CγH2, CδH2), 0.89 | ||||||||
| [t, 3H, 3J = 6.8, (CεH3)] | ||||||||
| 85.4 [dd, 1J(RhP2) = 184, | 69.8 (m) | 48.4 [d, 1J(RhC) = 12.7] | 30.8 | 57.6 [dd, 1J(PC) = | ||||
| 2J(PP) = 7.5] | 3.17 (m) | 1.82 (s) | 10.5, 2J(RhP) = 4.1] | |||||
| 4.45 [d, 2J(PH) = 10.0] | ||||||||
| 12d | 85.6 [dd, 1J(RhP1) = 179, | |||||||
| 2J(PP) = 7.4] | ||||||||
| 84.0 [dd, 1J(RhP2) = 182, | ||||||||
| 2J(PP) = 7.4] |
ia brown to
deep green and then to dark red. The alkali metal was removed
by filtration and the solution layered with 5 ml hexane.
The product was isolated as a deep red microcrystalline powder
(65 mg, 72%). Selected NMR data are given in Table 4.In the case of 12c a microcrystalline product was obtained directly from the reaction mixture after a few minutes.
Acknowledgements
This work was supported by the Swiss National Science Foundation. S. D. thanks the Gottlieb–Daimler and Carl–Benz Foundation for a grant.References and notes
- Neutral mononuclear d9-rhodium and iridium carbonyls can only be isolated by matrix techniques: (a) A. J. L. Hanlan and G. A. Ozin, Inorg. Chem., 1979, 18, 2091 and refs. cited therein CrossRef; (b) A. B. P. Lever, G. A. Ozin, A. J. L. Hanlan, W. J. Power and H. B. Gray, Inorg. Chem., 1979, 18, 2088 CrossRef CAS; (c) J. H. B. Chenier, M. Histed, J. A. Howard, H. A. Joly, H. Morris and B. Mile, Inorg. Chem., 1989, 28, 4114 CrossRef CASd10-rhodate and d10-iridate anions: (d) [Rh(CO)4]−: P. Chini and S. Martinengo, Inorg. Chim. Acta, 1969, 3, 21 Search PubMed; (e) [Ir(CO)4]−: L. Malatesta, G. Caglio and M. Angoletta, Chem. Commun., 1970, 532 Search PubMed; (f) [Rh(CO)2(PPh3)2]− and [Ir(CO)3(PPh3)]−: J. P. Collman, F. D. Vastine and W. R. Roper, J. Am. Chem. Soc., 1968, 90, 2282 and refs. cited therein Search PubMed; (g) [Rh(CO)(triphos)]−: G. G. Johnston and M. C. Baird, J. Organomet. Chem., 1986, 314, C51. Search PubMed.
- d10-M fluorophosphine complexes (M = Rh, Ir): (a) [Rh(PF3)4]−: T. Kruck, N. Derner and W. Lang, Z. Naturforsch. B, 1966, 21, 1020 Search PubMed; (b) [Ir(PF3)4]−: M. A. Bennett and D. J. Patmore, Inorg. Chem., 1971, 10, 2387 and refs. cited therein Search PubMed[Rh(PF2NMe2)4]−: D. A. Clement and J. F. Nixon, J. Chem. Soc., Dalton Trans., 1973, 195 RSC.
- (a) [Rh(Ph2P–(CH2)n–PPh2)2]− (n = 2, 3): B. Bogdanović, W. Leitner, C. Six, U. Wilczok and K. Wittmann, Angew. Chem., 1997, 109, 518 Search PubMedAngew. Chem., Int. Ed. Engl., 1997, 36, 500 Search PubMed (b) [Ir(dppf)2]− [dppf = 1,1′-bis(diphenylphosphino)ferrocene]: B. Longato, L. Riello, G. Bandoli and G. Pilloni, Inorg. Chem., 1999, 38, 2818 Search PubMed.
- (a) H. Schönberg, S. Boulmaâz, M. Wörle, L. Liesum, A. Schweiger and H. Grützmacher, Angew. Chem., 1998, 110, 1492 CrossRefAngew. Chem., Int. Ed., 1998, 37, 1423 Search PubMed (b) S. Boulmaâz, M. Mlakar, S. Loss, H. Schönberg, S. Deblon, M. Wörle, R. Nesper and H. Grützmacher, Chem. Commun., 1998, 2623 RSC; (c) S. Deblon, unpublished results. Further details will be provided upon request to the authors.
- (a) S. Oishi, J. Mol. Catal., 1987, 39, 225 CrossRef CAS; (b) E. Amouyal, in Homogeneous Photocatalysis, ed. M. Chanon, Wiley, New York, 1997, vol. 2, pp. 264–307 Search PubMed.
- J. Thomaier, S. Boulmaâz, H. Schönberg, H. Rüegger, A. Currao, H. Grützmacher, H. Hillebrecht and H. Pritzkow, New J. Chem., 1998, 21, 947 RSC.
- E. A. Halevi and R. Knorr, Angew. Chem., 1982, 94, 307 CAS; Angew. Chem., Int. Ed. Engl., 1982, 21, 288 Search PubMed; T. A. Albright, J.-K. Burdett, and M.-H. Whangbo, Orbital Interactions in Chemistry, Wiley, New York, 1985, p. 305 Search PubMed.
- The strongest deviation (49.7°) was observed by: (a) V. Di Noto, G. Valle, B. Zarli, B. Longato, G. Pilloni and B. Corain, Inorg. Chim. Acta, 1995, 233, 165 CrossRef CASWe thank the authors for bringing this value to our attention. Further distorted complexes: (b) [RhCl(CO)(PBu3t)2] has the structure of a flattened tetrahedron: P–Rh–P 162°, Cl–Rh–C: 150°: H. Schumann, M. Heisler and J. Pickardt, Chem. Ber., 1977, 110, 1020 Search PubMed; (c) R. L. Harlow, S. A. Westcott, D. L. Thorn and R. T. Baker, Inorg. Chem., 1992, 31, 323, and refs. cited therein CAS; (d) Compare also distortion angles in other bisphosphane-bisolefin complexes like [Rh(cod)(R,R-Me–BPE)]+ (φ = 24°), [Rh(cod)(R,R-Me–DuPhos)]+ (φ = 18°): M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125 Search PubMed; (e) [Ir(cod)(R,R-Me–DuPhos)]+ (φ = 18°): B. F. M. Kimmich, E. Somsook and C. R. Landis, J. Am. Chem. Soc., 1998, 120, 10115 Search PubMed; (f) See also: A. Miedaner, R. C. Haltiwanger and D. DuBois, Inorg. Chem., 1991, 30, 417 Search PubMed.
- G. N. Walker and A. R. Engle, J. Org. Chem., 1972, 37, 4294 CrossRef CAS.
- K. C. Pich, R. Bishop, D. C. Craig and M. L. Scudder, Aust. J. Chem., 1994, 47, 837 CAS.
- For clarity, the orientation of the methyl groups in A, B and C is not specified in Scheme 2. A comparable mechanism has been proposed for isomerisation reactions in zinc thiolates containing a Zn2(μ-N)(μ-S) four-membered ring: H. Grützmacher, M. Steiner, H. Pritzkow, L. Zolnai, G. Huttner and A. Sebald, Chem. Ber., 1992, 125, 2199.
- J. Thomaier, PhD Dissertation, University of Freiburg, Freiburg, Germany, 1996..
- The figures were produced with the facilities included in the program package WINGX: L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1 RSC.
- (a) P. A. Evans, J. E. Robinson and J. D. Nelson, J. Am. Chem. Soc., 1999, 121, 6761 CrossRef CAS; (b) P. A. Evans and J. D. Nelson, J. Am. Chem. Soc., 1998, 120, 5581 CrossRef CAS; (c) P. A. Evans and J. D. Nelson, Tetrahedron Lett., 1998, 39, 1725 CrossRef CAS; (d) R. Takeuchi and N. Kitamura, New J. Chem., 1998, 22, 659 RSC.
- For recent review on transition-metal-catalysed allylic alkylations see: B. M. Trost and D. L. Van Vranken, Chem. Re., 1996, 96, 395 Search PubMed.
- (a) [RhCl(PPh3)3]: D. C. Olson and W. Keim, Inorg. Chem., 1969, 8, 2028 Search PubMed; (b) L. Horner, K. Dickerhof and J. Mathias, Phosphorus Sulfur Relat. Elem., 1981, 10, 349 Search PubMed; (c) [MCl(dppe)2], [MX(dppe)(CO)n], and [M(dppe)2]+ (M = Rh, Ir): G. Pilloni, E. Vecchi and M. Martelli, J. Electroanal. Chem., 1973, 45, 483 Search PubMed; (d) G. Pilloni, G. Zotti and M. Martelli, Inorg. Chim. Acta, 1975, 13, 213 CrossRef CAS; (e) J. A. Sofranko, R. Eisenberg and J. A. Kampmeier, J. Am. Chem. Soc., 1979, 101, 1042 CrossRef CAS; (f) J. A. Sofranko, R. Eisenberg and J. A. Kampmeier, J. Am. Chem. Soc., 1980, 102, 1163 CrossRef CAS; (g) G. Pilloni, G. Zotti and M. Martelli, Inorg. Chem., 1982, 21, 1283 CrossRef CAS; (h) F. Morandini, G. Pilloni, G. Consiglio and A. Mezzetti, Organometallics, 1995, 14, 3418 CrossRef CAS; (i) [Rh(P(OiPr3)4]+: G. Pilloni, G. Zotti and S. Zecchin, J. Organomet. Chem., 1986, 317, 357 Search PubMed; (j) [M(PPh3)3(L)]+: G. Zotti, S. Zecchin and G. Pilloni, J. Electroanal. Chem., 1984, 175, 241 Search PubMed; (k) [MX(CO)(PPh3)2]: G. Schiavon, S. Zecchin, G. Pilloni and M. Martelli, J. Organomet. Chem., 1976, 121, 261 Search PubMed; (l) G. Schiavon, S. Zecchin, G. Pilloni and M. Martelli, J. Inorg. Nucl. Chem., 1977, 39, 115 Search PubMed; (m) G. Zotti, S. Zecchin and G. Pilloni, J. Organomet. Chem., 1983, 246, 61 CrossRef CAS; (n) [Rh(cod)2]+: J. Orsini and W. E. Geiger, J. Electroanal. Chem., 1995, 380, 83 Search PubMed.
- Compare, for example, [Fe(Cp)2]/[Fe(Cp)2]+ ΔEred/ox = +450 mV and [Fe(Cp*)2]/[Fe(Cp*)2]+ ΔEred/ox = −100 mV (Cp = cyclopentadienyl; Cp* = pentamethylcyclopentadienyl): P. Zanello in Ferrocenes, eds. A. Togni and T. Hayashi, VCH–Wiley, Weinheim, 1995, p. 317. Search PubMed.
- Simulation software for cyclic voltammograms, version 3.03: M. Rudolph, D. D. Reddy and S. W. Feldberg, Anal. Chem., 1994, 66, 589, distributed by Bioanalytical Systems, Inc., West Lafayette, IN 47906, USA. Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |