A study of substrate specificity of toluene dioxygenase in processing aromatic compounds containing benzylic and/or remote chiral centers
Vu P.
Bui
a,
Trond Vidar
Hansen†
a,
Yngve
Stenstrøm†
a,
Tomas
Hudlicky
*a and
Douglas W.
Ribbons
b
aDepartment of Chemistry, Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersity of Florida, Gainesille, FL 32611-7200, USA. E-mail: hudlicky@chem.ufl.edu
ersity of Florida, Gainesille, FL 32611-7200, USA. E-mail: hudlicky@chem.ufl.edu
bSFB Biocatalysis at the Institute of Organic Chemistry, Technical Uniersity of Graz, Stremayrgasse 16, A-8010 Graz, Austria. E-mail: sekretariat@orgc.tu-graz.ac.at
First published on 19th December 2000
Abstract
A series of substituted arenes containing remote chiral centers were screened as substrates for toluene dioxygenase (TDO). The absolute stereochemistry of the new metabolites was determined by chemical and spectroscopic correlation with synthetic standards. There was no evidence for kinetic resolution; enantiomers were indiscriminately processed by the enzyme to diastereomeric pairs, which were separable upon derivatization. Some of these new metabolites are useful as synthons for morphine synthesis. Full experimental details are reported for those new compounds stable to isolation and for derivatives of those that are unstable.
Introduction
In 1987, nearly 20 years after the report by Gibson et al. of enzymatic dihydroxylation of p-chlorotoluene,1 Ley et al. described the first application of cis-cyclohexadienediols in synthesis.2 Since then the number of syntheses taking full advantage of enzymatically introduced asymmetry has burgeoned, as has the number of reports of isolation and identification of new metabolites derived from biooxidation of aromatic compounds. The isolation of diol metabolites and their synthetic applications have been reviewed recently.3–15 The two latest reviews list structures of most of the metabolites produced to date,3,4 grouped by the corresponding class of arenes from which they originate. Fig. 1 summarizes the structural diversity available through this transformation, which still does not have an asymmetric chemical equivalent.16 Of the several hundred metabolites identified to date, only 21 have been employed in synthesis, most of which (ca. 90%) are the diols derived from benzene, chlorobenzene or bromobenzene.3 | ||
| Fig. 1 Structural diversity of substrates processed by toluene dioxygenase (TDO) and naphthalene dioxygenase (NDO). | ||
There are fewer than ten metabolites reported from the biooxidation of aromatic compounds bearing chiral substituents;
Gibson etal. described the first such oxidation, of 1-phenyl-1-ethanol, in 1973.17 Other reports of oxidations of 1-phenyl-1-ethanol and related substrates catalyzed by toluene dioxygenase (TDO) have generally been connected to studies of mono ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) s. dihydroxylation.18,19 A recent paper by Boyd et al.20 discusses the issue of hydroxylation and the ability of TDO to
recognize enantiomers of 1-phenyl-1-ethanol.
s. dihydroxylation.18,19 A recent paper by Boyd et al.20 discusses the issue of hydroxylation and the ability of TDO to
recognize enantiomers of 1-phenyl-1-ethanol.
We have published a preliminary report on the oxidation of several substrates having either a chiral center at a benzylic position or at other chiral centers located α or β to the benzene ring.21 We found no evidence of discrimination, with the exception of a slight preference for the oxidation of S-1-phenyl-1-ethanol when the blocked mutant22Pseudomonas putida 39D was used. No preference was detected in the oxidations mediated by TDO expressed in the recombinant organism Escherichia coli JM109 (pDTG601).23 Here, we report the experimental details and absolute stereochemistry correlations of the new metabolites.
Results and discussion
Our interest was focused primarily on phenyl-substituted cyclohexanols and cyclohexanones (Table 1) because of the relevance of these compounds to our synthetic approaches to morphine (23). With these cases, the issue of kinetic resolution of enantiomers is less important than the enzyme's acceptance of both enantiomers of the substrates, because it is ultimately our intention to re-aromatize the biooxidation products—by means of either E. coli JM109 (pDTG601), followed by chemical oxidation, or E. coli JM109 (pDTG602), an organism that expresses both TDO and catechol dehydrogenase (DHCD)24—to produce advanced morphine intermediates with the catechol unit already attached to ring A, Fig. 2.25| Substrate | Product | Substrate | Product | |
|---|---|---|---|---|
| a Numbers in brackets denote the isolated yield of metabolite in g L−1; yields are not optimized. b Detected by UV in shake-flask experiments only. | ||||
|

|

|

|
|
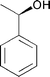
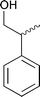
| 1a | 2a [1.0]a | 1b | 2b [1.0]a | |
| (±)-1 | 2a:2b = 1:1 (JM109) |

|
 b
b
|
|
| 2a:2b = 2:3 (Pp39D) [5.0]a | 3 | 4 | ||
|
 b
b
|

|
 b
b
|
|
| 5 | 6 | 7 | 8 | |

|

|

|

|
|
| (+)-9 | 10a [1.0]a | (−)-9b | 10b [1.0]a | |
| (±)-9 | 9a:9b = 1:1 (JM109) [1.2]a | |||

|

|

|
|

|
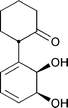
| (+)-11a | 12a | (−)-11b | 12b | 12c |
| (±)-11 | 12a:12b:12c = 2:1:1 (JM109) [1.0] |

|
 b
b
|
|
| 13 | 14 | |||

|
|
|
|
|
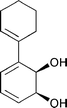
| 15 | 16 [3.0]a | 17 | 18 [1.8]a | |

|

|

|

|
|
| 19 | 20 [0.8]a | 21 | 22 [1.1]a | |
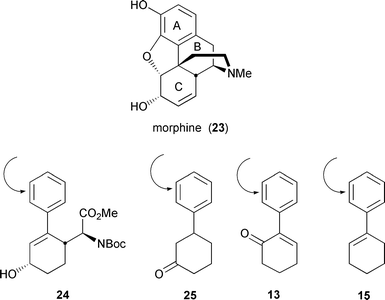 | ||
| Fig. 2 Introduction of the catechol unit to morphine synthons. | ||
If TDO actually discriminates enantiomers in more highly substituted cases, it would be useful for approaches to the syntheses of both enantiomers of morphine by kinetic resolution. However, biooxidation of enantiomers of the substrates does not appear to discriminate between enantiomers; it produces a mixture of diastereomers which, on separation, would also be useful in an enantiodivergent approach to the target. Nevertheless, for the time being, it was more important for us to probe the limits of substitution on the substrates and how the presence of chiral centers would be tolerated by the enzyme. That both substrates 9 and 11 were processed by TDO, albeit without enantiomeric preference, bodes well for attempting to generate the required catechol units from compounds such as 13, 15, or 25, as well as the relatively complex advanced intermediate 24.26
Trial fermentations were carried out in shake flasks and monitored by UV for the presence of the desired metabolites. On detection of a cyclohexadienediol, the process was scaled up in either a 10 or 15 L fermentor. Isolated compounds were subjected to rigorous structure proof, including absolute stereochemistry, as described in the Experimental. Pure enantiomers as well as racemic mixtures of substrates 1, 9 and 11 were subjected to oxidation by TDO.
In only one case was any enantiomeric preference detected—in the oxidation of (±)-1 with the blocked mutant22P. putida 39D, in agreement with an earlier observation by Ribbons and Ahmed.19 The use of recombinant clones did not reveal any preference for enantiomers, and we rationalized this by slight differences in the topology of the enzymes from the two organisms as a result of post-translational folding and structural modification. The substrates shown in Fig. 3 were not recognized by TDO in either of the organisms, nor were they recognized by naphthalene dioxygenase (NDO).21
 | ||
| Fig. 3 Substrates not oxidized by toluene dioxygenase (TDO) or naphthalene dioxygenase (NDO). | ||
Initially, we subjected compounds 28, 29 and 30 (Fig. 3) to both TDO and NDO because of the overlap in recognition of substrates by the two enzymes.21 Because none were oxidized, we also looked at a series of phenethyl alcohols related to 1 having ortho, meta and para methyl groups. Of these, only the para isomer was recognized by TDO, and this may explain why 28 and 30 were not processed by the enzyme. Differences in recognition rates for ortho, meta and para substitution have been studied previously on several compounds, for example halostyrenes and ring-halogenated ethylbenzenes,27–30 with the conclusion that ortho or meta substitution leads to lower yields of metabolites.
Structure and absolute stereochemistry of metabolites
Although the diols derived from 1-phenyl-1-ethanols (1a, 1b) are known,17–19 we carried out a potassium azodicarboxylate (PAD) reduction of racemic 2a,b to 31 (Scheme 1) and confirmed a 1:1 ratio of diastereomers by NMR. Diols 4, 6 and 8 were detected by UV (λmax = 262, 270 and 266 nm, respectively) only in shake-flask experiments and were not isolated. | ||
| Scheme 1 Reagents: i, PAD, AcOH, MeOH; ii, DMP, p-TsOH; iii, CBr4, PPh3; iv, Bu3SnH, AIBN; v, Mg, Et2O, Li2CuCl4; vi, cyclohexanyl triflate; vii, sec-BuLi, Me3SnCl; viii, (CH3CN)2PdCl2, cyclohexenyl triflate, DMF; ix, THF, H2O, TFA; x, sec-BuLi, BnCl; xi, NaBH4, EtOH, rt; xii, MeOH, (PPh3)3RhCl, H2, 30 psi. | ||
Substrates 5 and 7 were screened in order to probe the tolerance of the enzyme for increased size and functionality of
the aromatic substituent. We expected that the relatively
bulky nature of these substrates would lead to preferential
oxidation of one enantiomer; however, (±)-9 was oxidized to a
1:1 mixture of 10a and 10b, and (±)-11 to a 2:1:1 mixture of 12a, 12b and 12c. These observations confirmed that the enzyme did not discriminate enantiomers. Diols 10a and
10b were also reduced with PAD to 32a and 32b, respectively, which were subsequently converted to the diastereomeric bromides and reduced to 33, a compound that was independently synthesized (ia cuprate coupling with cyclohexyl triflate) from the vinyl bromide 34, whose absolute configuration is known.31 The two materials were identical in all respects, thus providing a proof of the absolute stereochemistry
of 10a and 10b. In addition, 18 was reduced to 35, protected as acetonide 33, and found to be identical to the material derived from 34 or 10a and 10b. The absolute configuration of 16 was similarly proven by conversion of 16 to diene 36, which was matched with the compound synthesized from
34ia its trimethylstannane derivative and a palladium-catalyzed coupling with an enol triflate derived from cyclohexanone (Scheme 1). Diol 14 (which we considered for its potential use in our approach to morphine, Fig. 2) was detected by UV (λmax = 266 nm) but was not isolated because of difficulties in scale-up.
To prove its absolute stereochemistry, diol 20 was subjected to PAD reduction with the expectation that both of the unhindered double bonds would be reduced to provide 35, a compound of known absolute stereochemistry; however, the reduction provided a mixture of inseparable diols 37 and 38. These two compounds were protected as acetonides and sub jected to catalytic hydrogenation with Wilkinson's catalyst to furnish compound 33, of known absolute stereochemistry, thus confirming the absolute stereochemistry of 20 (Scheme 1).
To establish the absolute stereochemistry of 22, the compound was reduced with PAD to diol 39, which was also chemically synthesized by coupling vinyl bromide 34 with benzyl chloride using sec-BuLi as the metal–halogen exchange reagent (Scheme 1). The biooxidation of 21 to diol 22 served as a background experiment for the attempted kinetic resolution study of 29, shown in Fig. 3.
Pure enantiomers of 11a and 11b, by pyridinium chlorochromate (PCC) oxidation of commercially available alcohols 9a and 9b, were each subjected to fermentation. The products were isolated and characterized, as shown in (Scheme 2).
 | ||
| Scheme 2 Reagents: i, PCC, CH2Cl2; ii, E. coli JM109 (pDTG601). | ||
It is clear that cyclohexyl-substituted arenes are good substrates for TDO, with both enantiomers being processed by the enzyme to afford the corresponding diasteromeric diols. Because some of the compounds contain structural elements of morphine, it will be worthwhile to investigate greater limits of substitution.
Conclusions
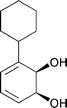
Several new diol metabolites were isolated and identified from arenes having chiral substituents. We found that TDO did not discriminate between enantiomers. Now, the more important issue with respect to morphine synthesis is the introduction of the catechol unit directly onto an aromatic ring, a process difficult to accomplish by chemical means when there are sensitive functionalities elsewhere. We have performed preliminary experiments with JM109 (pDTG602) on several functionalized aromatics and found that they can be cleanly converted to catechols.32 Of direct relevance to morphine synthesis was the successful oxidation of phenylcyclohexane, which led to the corresponding catechol33 (Scheme 3).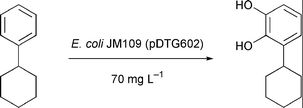 | ||
| Scheme 3 | ||
These endeavors were intended to identify substrates in which enantiomeric discrimination (by TDO), processing of both enantiomers to the corresponding diols (TDO), or direct introduction of a catechol unit (TDO and DHCD) could be achieved. The second topic will be expanded to other substrates useful in morphine synthesis, as diastereomeric cyclohexadienediols can be utilized separately in an enantiodivergent approach to the alkaloid. Further development in these areas will be reported in due course. The last project was successful and bodes well for direct biocatalytic synthesis of functionalized catechols. It will be investigated further for its potential in the environmentally benign synthesis of functionalized catechols.32
Experimental
General
1H and 13C NMR spectra were recorded at 300 and 75 MHz, respectively, on a Varian 300 spectrometer, unless otherwise stated. Chemical shifts are reported relative to TMS, CDCl3 or DMSO-d6. Coupling constants are measured in Hz. Multiplicities in 13C NMR are based on APT or DEPT. IR spectra were recorded on a Perkin-Elmer FT-IR (KBr) and are given as wavenumbers in cm−1. For the mass spectra, relative abundance is reported in parentheses. High resolution mass spectra and elemental analyses were performed at the University of Florida and at Atlantic Microlab, Inc, respectively. Optical rotations were recorded on a Perkin-Elmer 241 digital polarimeter (10−1 deg cm2 g−1). Melting points were obtained on a Thomas-Hoover capillary melting point apparatus and are uncorrected. Gas chromatographic analysis of enantiomeric mixtures was performed on a cyclodextrin column (Chirasil DexCB, 25 m × 0.32 mm) with helium as the carrier gas. Reversed-phase HPLC analyses were performed on a C18 column (Phenomenex 250 × 10 mm) using mixtures of water and acetonitrile as eluents. For chromatographic purification, silica gel (230–400 mesh) was used.General procedure for the large-scale (15 L) fermentation with Escherichia coli
A mineral salts broth (MSB, 600 mL) containing K2HPO4 (9.6 g), KH2PO4 (8.4 g), (NH4)2SO4 (3 g), yeast extract (9 g), glucose (18 g), and MgSO4·7H2O (1.2 g) was divided into two 2.8 L Fernbach flasks and sterilized. After the broth had cooled to ambient temperature, ampicillin (100 mg L−1) was added, and each flask inoculated with a single colony of E. coli JM109 (pDTG 601) from a fully grown plate. The inoculum was grown overnight on an orbital shaker (35 °C, 150 rpm) and added to sterilized production medium (8 L) in a 15 L fermentor. The production medium consists of an aqueous mixture of KH2PO4 (60 g), citric acid (16 g), MgSO4·7H2O (40 g), trace metal solution [16 mL: Na2SO4 (1 g L−1), MnSO4 (2 g L−1), ZnCl2 (2 g L−1), CoCl2 ·6H2O (2 g L−1), CuSO4·5H2O (0.3 g L−1), FeSO4·7H2O (10 g L−1), pH 1.0], conc. H2SO4 (9.6 mL) and ferric ammonium citrate (9.6 mL, 270 g L−1). The mixture was neutralized with conc. ammonia and supplemented with ampicillin (800 mg) and thiamine hydrochloride (2.69 g). The culture was grown for 24 h with concentrated glucose solution (720 g L−1) as the carbon source, then induced with 80 mg isopropyl β-D-thiogalactopyranoside (IPTG) after the optical density (OD) had reached at least 15%. Once the OD was 45% or higher, the cells were used for screening new substrates or large-scale fermentations.General procedure for screening of new substrates
1 L of the high OD culture was centrifuged (3000 rpm, 15 min). The cells were suspended in 300 mL of 0.20 M KPO4 buffer (pH 7.0) supplemented with 2% glucose. The substrate was added in small portions with the pH maintained at 7.0 with 10 M NaOH. The progress of the biotransformation was monitored by UV absorbance of the broth in the region 260–270 nm and by TLC. The substrates metabolized under these conditions were then subjected to large-scale biooxidation in a 15 L fermentor. (Compounds 4, 6, 8 and 14 were not scaled up.)General method for the PAD reduction of diene-diols
The crude fermentation product (10 mmol) was dissolved in cold methanol (5 mL), and the mixture cooled in an ice bath. Potassium azodicaboxylate (PAD) (4.9 g, 25 mmol) was added with stirring. A solution of acetic acid (4.6 mL, 80 mmol) in methanol (20 mL) was added dropwise to the reaction vessel at such a rate that 1–2 bubbles were produced per second. Acetic acid addition took 3–4 h. The progress of the reaction was monitored by UV or by 1H NMR for the disappearance of olefinic protons. The mixture was allowed to warm to ambient temperature, with the color changing from yellow to milky white. The mixture was stirred for 3 h, then saturated NaHCO3(aq) added until effervescence ceased and the methanol removed under reduced pressure. The residue was diluted with brine (10 mL), and the aqueous solution extracted with EtOAc (4 × 20 mL). The combined organic phases were dried (MgSO4) and concentrated to yield the reduced product.Biooxidation of (±)-1-phenyl-1-ethanol (1) with Pseudomonas putida 39D
The culture was prepared according to the procedure of Hudlicky et al.34 The culture was induced by adding 150–200 mg of toluene and shaken for 2–3 h, after which time the cells were centrifuged (3000 rpm, 10 min) and resuspended in the same nutrient broth (250 mL). Racemic 1 (300 mg) was added to the culture. The conversion was monitored by the increase of the UV absorption of the formed diene at 266 nm and by TLC (EtOAc). The biooxidation was allowed to proceed for another 58 h. The cells were removed by centrifugation, and the supernatant was extracted with EtOAc (5 × 250 mL). The organic layers were combined, dried over Na2SO4 and concentrated to give 138 mg of residue, which was analyzed by GLC on an optically active cyclodextrin column, showing it to be a mixture of dienediols and starting material. Comparison to standard samples showed the residual 1-phenylethanol to be a 60:40 mixture of the R- and the S-enantiomers, respectively.Biooxidation of (R)-1-phenyl-1-ethanol (1a) and (S)-1-phenyl-1-ethanol (1b)
Biooxidation of each enantiomer was carried out as described for racemic 1-phenyl-1-ethanol with P. putida 39D. The conversion was monitored by UV spectroscopy (λmax = 270 nm). (R)-1-Phenyl-1-ethanol (1a) yielded 3-[1(R)-hydroxyethyl]cyclohexa-3,5-diene-1(S),2(R)-diol (2a) and (S)-1-phenyl-1-ethanol (1b) yielded 3-[1(S)-hydroxyethyl]cyclohexa-3,5-diene-1(S),2(R)-diol (2b) after respective work-up. In each case, the yield was 1 g L−1.Biooxidation of (±)-1-phenyl-1-ethanol (1) with Escherichia coli JM109 (pDTG601)
The cultures were prepared according to the procedure of Hudlicky et al.,30,34 and 1.0 g of substrate was added to the cell culture. The progress was monitored by UV absorbance (λ = 266 nm). GLC analysis revealed a 1:1 mixture of diastereomers 2a and 2b. The 1H NMR spectrum was in agreement with that published in the literature.17 The biooxidation of racemic 1-phenyl-1-ethanol was also carried out on a large scale to afford a crude yield of 40 g (5 g L−1) of a 1:1 mixture of as reddish oil.Biooxidation of (±)-1-(4-methylphenyl)-1-ethanol (3), (±)-2-phenyl-1-propanol (5) and (±)-1-phenyl-2-propyn-1-ol (7)
The biooxidations of 3, 5 and 7 with E. coli JM109 (pDTG601) were carried out as described for the small scale transformation for racemic 1-phenyl-1-ethanol (1). The progress of the biotransformations was monitored by UV absorbance (λmax = 262–272 nm). The metabolites 4, 6 and 8 were detected by UV only, with λmax of 262, 270 and 266 nm, respectively, and were not isolated.Biooxidation of (1S,2R)-(+)-trans-2-phenyl-1-cyclohexanol (9a)
The biooxidation of 9a with E. coli JM109 (pDTG601) was carried out as described for the small-scale transformation of racemic 1-phenyl-1-ethanol (1). The progress of the biotransformations was monitored by UV absorbance (λ = 270–272 nm) and TLC (EtOAc). The metabolite 10a was isolated according to the method described for that of (±)-1-phenyl-1-ethanol. The crude material was immediately subjected to PAD reduction. Purification by flash chromatography (1:4 hexane–EtOAc) afforded 100 mg (49%) of 32a.(1S,2R,1′R,2′S)-3-(2′-Hydroxycyclohexanyl)-3-cyclohexene-1,2-diol (32a)
R f = 0.68 (EtOAc); mp = 89–91 °C; [α]D28 −82.3 (c 0.6, CH2Cl2); IR (film): 3329, 1447 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 5.50 (m, 1H), 4.83 (d, J = 4.5, 1H), 4.73 (d, J = 3.4, 1H), 4.30 (d, J = 6.1, 1H), 3.73 (t, J = 3.9 Hz, 1H), 3.46–3.38 (m, 1H), 3.26–3.15 (m, 1H), 2.1–1.0 (m, 13H); 13C NMR (75 MHz, CDCl3): δ 139.9, 125.5, 75.0, 69.9, 69.5, 49.9, 35.3, 32.3, 25.9, 25.1, 24.8, 24.2; MS (CI +): m/z 213 (22), 195 (100), 177 (87), 159 (29), 147 (59), 105 (24); HRMS: calc. for C12H21O3, 213.1490. Found 213.1456. Anal. calc. for C12H20O3: C, 67.89; H, 9.50%. Found: C, 67.60; H, 9.55%.Biooxidation of (1R,2S)-(−)-trans-2-phenyl-1-cyclohexanol (9b)
The biooxidation of 9b with E. coli JM109 (pDTG601) was carried out as described for the small-scale transformation for racemic 1-phenyl-1-ethanol (1). The progress of the biotransformations was monitored by UV absorbance (λ = 270 nm) and TLC (EtOAc). The crude metabolite 10b was reduced with PAD and purified by flash chromatography (1:4 hexane–EtOAc) afforded 102 mg (50%) of 32b.(1S, 2R, 1′S, 2′R)-3-(2-hydroxycyclohexanyl)-3-cyclohexene- 1,2-diol (32b)
R f = 0.68 (EtOAc); mp = 103–106 °C; [α]D31 −148.0 (c 1, CH2Cl2); IR (film): 3364, 1448 cm−1; 1H NMR (300 MHz, CDCl3): δ 5.70 (dd, J = 2.6, 4.5, 1H), 4.16 (d, J = 3.3 Hz, 1H), 3.70–3.60 (m, 2H), 3.10 (bs, 1H), 2.85 (bs, 1H), 2.30–2.00 (m, 5H), 1.90–1.62 (m, 4H), 1.58–1.50 (m, 1H), 1.41–1.20 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 138.9, 129.5, 72.1, 70.7, 65.8, 54.1, 34.5, 32.2, 25.9, 25.3, 25.2, 24.7; MS (CI +): m/z 213 (29), 195 (78), 177 (100), 159 (28), 147 (16), 105 (14); HRMS: calc. for C12H21O3, 213.1490. Found: 213.1511. Anal. calc. for C12H20O3: C, 67.89; H, 9.50%. Found: C, 67.78; H, 9.62%.Biooxidation of (±)-trans-2-phenyl-1-cyclohexanol (9)
Into a 12 L fermentor with fully grown cells (OD640nm = 45), (±)-trans-2-phenyl-1-cyclohexanol (10.29 g) was added (2 g every 30 min). The progress of the biotransformation was monitored by UV absorbance (λ = 270 nm). Three hours after the first addition, the absorbance at 270 nm ceased to increase. The pH of the medium was adjusted to 7.8 with conc. NH4OH. Cells were removed by centrifugation, and the supernatant was extracted with base-washed ethyl acetate to afford 10.25 g (1.3 g L−1) of a gray solid as a 1:1 mixture of inseparable diastereomers of 3-(2-hydroxycyclohexanyl)-3,5-cyclohexadien-1,2-diol (10a and 10b).(R)- and (S)-2-Phenylcyclohexanone (11a and 11b)
11a and 11b were made by oxidizing the corresponding optically active trans-2-phenylcyclohexanol with pyridinium chlorochromate (PCC) in CH2Cl2 at 5 °C with the usual work-up.(R)-2-Phenylcyclohexanone (11a)
R f = 0.30 (hexane–Et2O 4:1); mp 48–51 °C; [α]D27 +108.7 (c 0.20, benzene), lit.35 [α]D26 +114.7 (c 0.45, benzene); IR (KBr): 3090, 1699 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.34–7.16 (m, 5H), 3.62 (dd, J = 12.0, 5.5 Hz, 2H), 2.55–2.38 (m, 2H), 2.27–2.22 (m, 1H), 2.14–2.08 (m, 1H) 2.02–1.95 (m, 2H), 1.85–1.77 (m, 2H); 13C NMR (300 MHz, CDCl3): δ 210.2, 138.8, 128.8, 128.5, 126.7, 57.7, 42.0, 35.3, 27.6, 25.0.(S)-2-Phenylcyclohexanone (11b)
[α]D27 −109.8 (c 0.30, benzene), lit.35 [α]D24 −113.5 (c 0.60, benzene); IR (KBr): 3092, 1695 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.36–7.15 (m, 5H), 3.62 (dd, J = 12.0, 5.5 Hz, 2H), 2.56–2.39 (m, 2H), 2.28–2.24 (m, 1H), 2.13–2.08 (m, 1H) 2.02–1.95 (m, 2H), 1.85–1.78 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 210.2, 138.8, 128.8, 128.5, 126.7, 57.7, 42.0, 35.3, 27.6, 25.0.Biooxidation of (±)-2-phenylcyclohexanone (11a and 11b)
Biooxidation of 11a and 11b with E. coli JM109 (pDTG601) was carried out as described for the small-scale transformation of racemic 1-phenyl-1-ethanol (1). This afforded the acetals 12a and 12c along with the ketone 12b in a 2:1:1 mixture, respectively.The proof of structures and the correlation of absolute stereochemistry was provided by transforming the two enantiomers 11a and 11b separately, as for racemic 11.
(4S,4aR,9aR)-4,6,7,8,9,9a-Hexahydro-4aH-dibenzofuran-4,5a-diol (12a)
λ max = 266 nm; Rf = 0.58 (1:1 hexane–EtOAc); [α]D28 +130.3 (c 0.7, MeOH); IR (film): 3378, 1449 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.09 (dd, J = 4.8, 9.4, 1H), 5.98 (dd, J = 4.3, 9.4, 1H), 5.84–5.78 (m, 1H), 4.76–4.70 (m, 1H), 4.25 (bs, 1H), 4.12 (bs, 1H), 3.97 (t, J = 5.5 Hz, 1H), 2.47–2.38 (m, 1H), 2.18–2.06 (m, 1H), 1.83–1.74 (m, 1H), 1.62–1.56 (m, 3H), 1.48–1.08 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 144.3, 127.8, 124.2, 114.9, 106.9, 79.7, 61.6, 49.1, 33.9, 30.2, 24.0, 22.8; MS (CI): m/z 192 (12), 191 (40), 132 (32), 128 (35), 119 (32), 107 (31), 105 (29), 91 (100), 83 (21), 81 (37); HRMS (CI) m/z calc. for C12H13O2 (M − OH): 191.1072. Found: 191.1090.(1S,5′S,6′R)-5′,6′-Dihydroxybicyclohexyl-1′,3′-dien-2-one (12b)
R f = 0.42 (hexane–EtOAc 1:1); [α]D28 +58.1 (c 0.6, MeOH); IR (film): 3352, 1702, 1444 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.06 (dd, J = 4.7, 9.5, 1H), 5.95 (dd, J = 5.6, 9.5, 1H), 5.81–5.77 (m, 1H), 4.74–4.69 (m, 1H), 4.20 (bs, 2H), 3.95 (t, J = 5.6 Hz, 1H), 2.45–2.38 (m, 1H), 2.14–2.04 (m, 1H), 1.80–1.72 (m, 1H), 1.68–1.58 (m, 3H), 1.46–1.12 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 213.2, 139.2, 129.2, 124.0, 114.6, 68.7, 68.5, 42.1, 30.1, 27.3, 25.9, 22.8; HRMS (CI): m/z calc. for C12H17O3 (M + H): 209.2675. Found: 209.2669.(4S,4aR,9aS)-4,6,7,8,9,9a-Hexahydro-4aH-dibenzofuran-4,5a-diol (12c)
λ max = 266 nm; Rf = 0.49 (hexane–EtOAc; 1:1); [α]D28 −126.3 (c 0.5, MeOH); IR (film): 3371, 1454 cm−1; 1H NMR (300 MHz, CDCl3): δ (6.05 (dd, J = 4.8, 9.4, 1H), 5.92 (dd, J = 4.3, 9.4, 1H), 5.80–5.75 (m, 1H), 4.72–4.68 (m, 1H), 4.20 (bs, 1H), 4.10 (bs, 1H), 3.98 (t, J = 5.5 Hz, 1H), 2.48–2.40 (m, 1H), 2.17–2.07 (m, 1H), 1.85–1.77 (m, 1H), 1.60–1.54 (m, 3H), 1.45–1.10 (m, 3H); 13C NMR (75 MHz, CDCl3): δ 144.8, 127.8, 124.4, 114.4, 105.7, 79.7, 61.7, 49.0, 33.8, 30.1, 24.2, 22.7; HRMS (CI): m/z calc. for C12H13O2 (M − OH): 191.1072. Found: 191.1081.Biooxidation of 2-phenyl-2-cyclohexenone (13)36
The biooxidation of 13 with E. coli JM109 (pDTG601) was carried out as described for the small-scale transformation of racemic 1-phenyl-1-ethanol (1). The progress of the reaction was monitored by UV (λ = 266 nm). The metabolite (14) was detected by UV only and was not isolated.Biooxidation of 1-phenylcyclohexene (15)
The biooxidation of 15 was performed according to the general procedure for the large-scale fermentation. Addition of 1-phenylcyclohexene (15.7 g, 99.2 mmol) afforded a crude yield of 18.4 g (3 g L−1) of 16 as an off-white solid.(1S,2R)-3-(1-Cyclohexenyl)-3,5-cyclohexadien-1,2-diol (16)
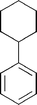
λ max = 302 nm; mp 90–93 °C; [α]D28 +47.5 (c 0.5, MeOH); IR (KBr): 3226, 3050, 1435 cm−1; 1H NMR (300 MHz, CDCl3): δ 6.16 (bt, J = 4.2, 1H), 6.01–5.89 (m, 2H), 5.75 (bd, J = 9.5, 1H), 4.49–4.43 (m, 1H), 4.34 (bd, J = 5.5 Hz, 1H), 2.70 (bs, 2H), 2.27–2.14 (m, 4H), 1.79–1.55 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 138.8, 133.8, 130.7, 125.7, 124.0, 117.9, 71.4, 66.7, 26.0, 25.3, 22.6, 22.0; MS (CI): m/z 193 (1), 191 (27), 175 (100), 174 (79), 173 (22); HRMS (CI): m/z calc. for C12H17O2 (M + H): 193.1228. Found: 193.1213.Biooxidation of phenylcyclohexane (17)36
The biooxidation of 17 was performed according to the general procedure for the small-scale fermentation. Addition of phenylcyclohexane (26.5 g, 165 mmol) afforded 14.0 g (1.8 g L−1) of 18 as an off-white solid.(1S,2R)-3-(1-Cyclohexyl)-3,5-cyclohexadien-1,2-diol (18)
R f = 0.32 (hexane–EtOAc 1:1); mp = 63–65 °C; λmax = 268 nm; [α]D30 −73.8 (c 1.0, CH2Cl2); IR (KBr): 3312, 3048, 1646, 1588, 1447 cm−1; 1H NMR (DMSO-d6): δ 5.80 (ddd, J = 2.3, 5.4, 9.5, 1H), 5.62–5.56 (m, 2H), 4.67 (d, J = 6.4, OH), 4.30 (d, J = 6.3, OH), 4.08–4.00 (m, 1H), 3.75 (bt, J = 5.4 Hz, 1 H), 2.10–1.98 (m, 1 H), 1.86–1.60 (m, 5 H), 1.34–0.96 (m, 5 H); 13C NMR (DMSO-d6): δ 146.9, 129.5, 123.0, 116.7, 69.7, 67.0, 41.4, 32.8, 31.2, 26.3, 26.2, 25.9.Biooxidation of 3-phenylcyclohexene (19)37
The biooxidation of 19 was performed according to the general procedure for the small-scale fermentation. Addition of 3-phenylcyclohexene (500 mg) afforded a crude yield of 120 mg of 20 as a reddish oil.(1S,2R)-3-(2-Cyclohexenyl)-3,5-cyclohexadien-1,2-diol (20)
R f = 0.53 (hexane–EtOAc 2:3); λmax = 262 nm; 1H NMR (DMSO-d6): δ (5.86–5.75 (m, 4H), 5.66–5.51 (m, 6H), 4.64 (d, J = 5.97, 2H), 4.41 (d, J = 6.36, 2H), 4.05 (bs, 2H), 3.78 (t, J = 5.97 Hz, 2H), 2.95 (bs, 2H), 1.98–1.91 (m, 4H), 1.86–1.70 (m, 2H), 1.60–1.40 (m, 6H).Biooxidation of diphenylmethane (21)
The biooxidation of 21 was carried out according to the general procedure for large-scale production. Diphenylmethane (17 g, 101 mmol) was slowly added to a fully grown (OD = 65) culture (8 L). Progress was monitored by UV (λmax = 266 nm). The biooxidation afforded 22 (8.8 g, 1.1 g L−1) as an off-white solid.(1S,2R)-3-Benzyl-3,5-cyclohexadien-1,2-diol (22)
R f = 0.47 (hexane–EtOAc 2:3); mp = 69–71 °C; [α]D33 +117 (c 1.0, CH2Cl2); IR (film): 3440, 3054, 2987, 1643, 1422, 1265, 896, 745, 705 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.34–7.26 (m, 2H), 7.23–7.16 (m, 3H), 5.85–5.75 (m, 1H), 5.68–5.60 (m, 1H), 5.54 (d, J = 4.43, 1H), 4.64 (d, J = 6.36, 1H), 4.52 (d, J = 6.17, 1H), 3.70 (t, J = 6.16 Hz, 1H), 3.46 (s, 2H); 13C NMR (75 MHz, CDCl3): δ 141.3, 139.1, 129.2, 128.7, 128.2, 126.6, 124.9, 121.1, 69.9, 69.3, 40.3; MS (FAB): m/z 202 (12), 185 (56), 184 (14), 107 (14), 93 (31), 91(100); HRMS (CI): m/z calc. for C13H14O2: 202.0994. Found: 202.0997.PAD reduction of racemic 2 to 31
The diasteromeric mixture of 2 was reduced with PAD according to the general procedure to afford 31 as a colorless oil.3-(Hydroxyethyl)cyclohexa-3-ene-1(S),2(R)-diol (31)
1H NMR (300 MHz, CDCl3): δ 6.00 (bt, J = 3.5, 1H), 5.89 (bt, J = 3.7 Hz, 1H), 4.47–4.42 (m, 1H), 4.40–4.26 (m, 4H), 4.17–3.90 (m, 5H), 3.76–3.52 (m, 2H), 2.30–2.12 (m, 2H), 2.11–1.90 (m, 2H), 1.85–1.56 (m, 4H), 1.36–1.20 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 140.3, 139.2, 126.5, 124.9, 71.0, 69.6, 69.2, 69.1, 67.6, 65.8, 24.9, 24.7, 23.9, 23.4, 21.8, 21.6.Correlation of absolute stereochemistry of 10a and 10b
The crude mixture from the biooxidation of (±)-9 was reduced with PAD to afford 32a and 32b. Protection of the mixture of theic-cis-diols 32a and 32b (390 mg, 2.0 mmol) was carried out in neat dimethoxypropane with a catalytic amount of p-toluenesulfonic acid to yield the corresponding ketals.
The crude mixture (378 mg, 1.5 mmol) was dissolved in dry CH2Cl2
(10 mL) at 0 °C and CBr4 (746 mg, 2.25 mmol) was added followed by PPh3 (393 mg, 2.25 mmol) portionwise over 10–15 min. The mixture was left for another two hours at ambient
temperature. Chromatography (2.5% EtOAc in hexane) yielded the two diastereomeric bromides (441 mg, 90%) which were reduced in the general manner using Bu3SnH and AIBN in dry, refluxing THF, affording crude 33 (377 mg, 80%). Purification by chromatography (EtOAc–hexane 1:9)
yielded an analytical sample with all spectroscopic and physical data in accord with 33 obtained from the synthetic approach described below. [α]D28
+49.6 (c 0.5, CHCl3).
Synthesis of (2,2-dimethyl-3a,7a-dihydrobenzo[1,3]dioxol-4-yl)cyclohexane (33)
The procedure for coupling was adapted from a report by Tamura and Kochi.38 Magnesium (98 mg, 4.0 mmol) was covered with 5 mL dry Et2O in a 100 mL 3-necked round-bottomed flask. The mixture was degassed with ultrasound under argon for 30 min. 7-Bromo-2,2-dimethyl-3a,4,5,7a-tetrahydrobenzo[1,3]dioxole (34) (0.70 g, 3.0 mmol) was added together with dry Et2O (5 mL) and left in the ultrasound bath for 1 h. The reaction mixture was then cooled to −78 °C, and with rigorous stirring Li2CuCl4 in Et2O (30 mL, 0.1 M) was added over 15 min. Stirring was continued for another 30 min, then cyclohexyl triflate (390 mg, 2.0 mmol) in dry Et2O (10 mL) was added. The reaction mixture was stirred for 3 h. Work-up in the usual manner followed by chromatography (5% EtOAc in hexane) afforded 18 mg (38%) of the product. [α]D27 +49.2 (c 0.9, CHCl3); 1H NMR (300 MHz, CDCl3): δ 5.52 (t, J = 3.0, 1H), 4.39 (d, J = 5.6, 1H), 4.22–4.17 (m, 1H), 2.18–2.04 (m, 1H), 2.02–1.93 (bt, J = 11.3 Hz, 1H), 1.87–1.54 (m, 7H), 1.35 (s, 6H), 1.34–0.93 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 141.2, 122.9, 107.9, 73.6, 72.9, 40.8, 33.3, 31.5, 28.0, 26.8, 26.6, 26.5, 26.4, 25.6, 20.7; MS (FAB): m/z 236 (0.6), 207 (40), 191 (21), 177 (12), 161 (18), 153 (42), 147 (100), 133 (42), 105 (21), 95 (35), 91 (63), 83 (70); HRMS (FAB): m/z calc. for C15H24O2: 236.1776. Found: 236.1774.Correlation of absolute stereochemistry of 12a and 12b
The crude mixture from the biooxidation of (±)-11 was treated with NaBH4 in MeOH to afford a mixture of diastereomeric alcohols 10, then the vicinal cis diol was protected as its acetonide with dimethoxypropane and cat. p-toluenesulfonic acid. This mixture was reduced with PAD according to the general procedure to yield 32. The crude product (800 mg, 3 mmol) was dissolved in dry CH2Cl2 (25 mL) at 0 °C and CBr4 (1.5 g, 4.5 mmol) was added, followed by portionwise addition of PPh3 (790 mg, 4.5 mmol) over 15 min. The mixture was stirred for 2 h at ambient temperature. Chromatography (2.5% EtOAc in hexane) yielded 700 mg (85%) of the two diastereomeric bromides, which were subsequently reduced with Bu3SnH and AIBN in dry THF at reflux to afford crude 33 (589 mg, 72%). Purification by chromatography (EtOAc–hexane 1:9) yielded an analytical sample whose spectroscopic and physical data were identical to those of synthetic 33 {[α]D28 +50.1 (c 0.6, CHCl3)}.Correlation of absolute stereochemistry of (18)
The cis-diene diol 18 was reduced with PAD according to the general procedure. Purification by flash chromatography (hexane–EtOAc 1: 4) afforded 35 as a crystalline compound (0.98 g, 86%).(1S,2R)-3-(1-Cyclohexyl)-3-cyclohexene-1,2-diol (35)
R f = 0.35 (hexane–EtOAc 1:1); mp = 68–69 °C; [α]D28 −75.2 (c 1.1, CH2Cl2); IR (KBr): 3330, 1448 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 5.34 (t, J = 3.5, 1H), 4.31 (d, J = 5.9, 1H, OH), 4.27 (d, J = 5.6, 1H, OH), 3.80 (t, J = 4.5 Hz, 1H), 3.45–3.33 (m, 1H), 2.12–1.85 (m, 3H), 1.82–1.52 (m, 6H), 1.49–1.39 (m, 1H), 1.32–0.90 (m, 5H); 13C NMR (75 MHz, DMSO-d6): δ 143.8, 120.9, 69.4, 67.0, 41.5, 38.0, 31.6, 26.6, 26.4, 26.0, 24.9, 24.2; MS (CI): m/z 195 (3, M − 1), 179 (100), 161 (18), 93 (33); HRMS (CI): m/z calc. for C12H19O (M − OH): 179.1436. Found: 179.1436. Anal. calc. for C12H20O2: C, 73.43; H, 10.27%. Found: C, 73.46; H, 10.38%.The spectroscopic and physical data of the acetonide of 35 were identical to those of synthetic 33.
Correlation of absolute stereochemistry of 20
The crude metabolites (7.5 g, 39 mmol) from a large-scale biooxidation of 19 were subjected to PAD reduction according to the general procedure to afford a mixture of inseparable diols 37 and 38. The crude mixture of products were protected as their acetonides, then subjected to catalytic hydrogenation (30 psi, MeOH, Wilkinson's catalyst). The product was purified by flash chromatography to afford a colorless oil (2.1 g) whose spectroscopic and physical data {[α]D28 +52.8 (c 0.6, CHCl3)} were in agreement with those of synthetic 33.Correlation of absolute stereochemistry of (1S,2R)-3-benzyl-3,5-cyclohexadien-1,2-diol (22)
In a flame-dried 25 mL round-bottomed flask equipped with a 3-way stopper, vinyl bromide 34 (172 mg, 0.746 mmol) was dissolved in freshly distilled THF (10 mL). The reaction vessel was cooled to −78 °C, then sec-BuLi (0.86 mL, 1.1 mmol) was added dropwiseia syringe. The mixture was stirred at −78 °C for
45 min, then benzyl bromide (0.19 g, 1.1 mmol) was added. The reaction was allowed to proceed for 1 h at −78 °C, then
the mixture was allowed to warm slowly to room temperature. The reaction was then quenched with 10% aq NH4Cl (5
mL) and ethyl acetate (10 ml) added. The layers were separated and the organic layer was washed with water (2 × 10 mL), then dried with MgSO4. The solvent was removed under
vacuum to afford the crude product (123 mg, 68%) as a light yellow oil. The crude product was dissolved in 6 mL of a 4:1:1 mixture of THF–H2O–TFA and allowed to stir at room temperature for 1 h. The mixture was concentrated under vacuum and the residue diluted with ethyl acetate (10 mL),
then washed with 5% NaHCO3(aq) (2 × 5 mL). The organic layers
were combined, dried over Na2SO4, and concentrated.
The product was purified by flash chromatography to afford
a white solid (84 mg, 82%). Physical and spectroscopic data
for the compound obtained from this route matched those of the
PAD-reduced diol 39 obtained from the biooxidation.
Synthesis of (1S,2R)-3-(1-cyclohexenyl)-3-cyclohexene-1,2-diol (36)
Into a flame-dried 50 mL round-bottomed flask equipped with a 3-way stopper, (CH3CN)2PdCl2 (5 mg, 0.019 mmol) and anhydrous DMF (5 mL) were added. A solution of cyclohexanone enol triflate39 (216 mg, 0.94 mmol) in dry DMF (5 mL) was added to the reaction flaskia syringe, under argon atmosphere. The trimethyltin derivative of the vinyl bromide 34 (300 mg, 0.94 mmol), prepared according to the procedure of Stille,40
was dissolved in dry DMF (5 mL), and the solution added dropwise to the reaction flask ia syringe. The starting material was consumed after 4 h of stirring at room temperature, according to TLC. The reaction mixture was washed with
5% NaHCO3(aq) (2 × 10 mL), and the aqueous layer was extracted several times with pentane. The organic layers were combined, dried with Na2SO4, and the solvent was removed
under vacuum to afford a red oil. The residue was dissolved in a solvent mixture of THF–H2O–TFA (4:1:1; 10 mL) and stirred for 1 h. The mixture was concentrated under vacuum and the residue washed with 5% NaHCO3 (2 × 5 mL) and water (2 × 5 mL). The organic layer was dried over Na2SO4 and the product purified by flash chromatography (hexane–EtOAc
1:4) to afford 36 as an off-white solid (43 mg, 18%). Rf = 0.54; mp = 143–144 °C;
[α]D28
−88.8 (c 1.0, CHCl3); IR (KBr): 3222,
3060, 1451 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 5.99 (m,
1H), 5.64 (m, 1H), 4.41 (d, J = 6.1, 1H), 4.24 (d, J = 5.2 Hz, 1H), 4.12 (m, 1H), 3.44–3.33 (m, 1H), 2.19–1.98 (m, 6H), 1.75–1.38 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 137.6, 134.3, 124.3, 123.5, 70.0, 65.9, 25.84, 25.77, 24.94, 24.85, 22.87, 22.21;
MS (FAB): m/z 194 (5), 177 (22), 93 (100); HRMS (CI): m/z calc. for C12H18O2 (M+): 194.1307. Found: 194.1253.
(1S,2R)-3-Benzyl-cyclohex-3-ene-1,2-diol (39)
An aliquot of the crude extract (2 g, 10 mmol) from biooxidation of substrate 21 was subjected to PAD (5.8 g, 30 mmol) reduction according to the general procedure. The product was purified by chromatography (hexane–EtOAc 1:1) to afford a white solid (0.98 g, 49%): mp = 73.5–75 °C; [α]D26 −160.4 (c 1.0, MeOH); IR (KBr): 3281, 3024, 1602, 1494, 1453, 1260, 1078 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.32–7.23 (m, 2H), 7.21–7.12 (m, 3H), 5.36 (s, 1H), 4.47 (d, J = 5.7, 1H), 4.31 (dd, J = 5.7, 1H), 3.62 (t, J = 4.5 Hz, 1H), 3.44–3.31 (m, 3H), 2.11–2.08 (m, 2H), 1.70–1.52 (m, 1H), 1.50–1.38 (m, 1H); 13C NMR (75 MHz, CDCl3): δ 139.7, 137.4, 129.2, 128.6, 127.2, 126.4, 69.8, 68.2, 41.0, 25.5, 24.1; MS (FAB): m/z: 188 (12), 187 (100), 185 (26), 169 (19), 93 (31), 91(38); HRMS (CI): m/z calc. for C13H16O2: (M − OH): 187.1123. Found: 187.1125. Anal. calc. for C13H16O2: C, 76.44; H, 7.90%. Found: C, 76.20; H, 7.93%.Acknowledgements
We are grateful to Professor David Gibson (University of Iowa) for providing us with a sample of E. coli JM 109 (pDTG602). Financial support from NSF (CHE-9615112 and CHE-9910412), EPA (R826113) and TDC Research, Inc., as well as from the Agricultural University of Norway (T. V. H. and Y. S.) and the Norwegian Research Council (T. V. H.) is gratefully acknowledged.References and notes
- D. T. Gibson, J. R. Koch, C. L. Schmid and R. E. Kallio, Biochemistry, 1968, 7, 3795 CrossRef CAS.
- V. Ley, F. Sternfeld and S. Taylor, Tetrahedron Lett., 1987, 28, 225 CrossRef.
- T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichim. Acta, 1999, 32, 35 Search PubMed.
- D. R. Boyd and G. N. Sheldrake, Nat. Prod. Rep., 1998, 309 RSC.
- S. M. Brown and T. Hudlicky, in Organic Synthesis: Theory and Applications, ed. T. Hudlicky, JAI Press, Greenwich, CT, 1993, vol. 2, p. 113. Search PubMed.
- D. A. Widdowson, D. W. Ribbons and S. D. Thomas, Janssen Chim. Acta, 1990, 8, 3 Search PubMed.
- H. A. J. Carless, Tetrahedron: Asymmetry, 1992, 3, 795 CrossRef CAS.
- G. N. Sheldrake, in Chirality and Industry, ed. A. N. Collins, G. N. Sheldrake and J. Crosby, John Wiley and Sons, Chichester, UK, 1992, p. 127. Search PubMed.
-
T. Hudlicky and J. W. Reed, in Adances in Asymmetric Synthesis, ed. A. Hassner, JAI Press, Greenwich, CT, 1995, p. 271. Search PubMed.
- A. D. Grund, SIM News, 1995, 45, 59 Search PubMed.
- T. Hudlicky and A. J. Thorpe, Chem. Commun., 1996, 1993 RSC.
- T. Hudlicky, Chem. Re., 1996, 96, 3 Search PubMed.
- T. Hudlicky, D. A. Entwistle, K. K. Pitzer and A. J. Thorpe, Chem. Re., 1996, 96, 1195 Search PubMed.
- T. Hudlicky, ACS Symp. Ser., 1996, 626, 180 Search PubMed.
- T. Hudlicky, in Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes, ed. P. T. Anastas and T. C. Williamson, Oxford University Press, Oxford, UK, 1998, ch. 10, p. 166. Search PubMed.
- A racemic bis-hydroxylation of benzene with OsO4 under protolytic conditions has been reported: W. B. Motherwell and A. S. Williams, Angew. Chem., Int. Ed. Engl., 1995, 34, 2031 CrossRef.
- D. T. Gibson, B. Gschwendt, W. K. Yeh and V. M. Kobal, Biochemistry, 1973, 12, 1520 CrossRef CAS.
- R. E. Cripps, P. W. Trudgill and G. Whately, Eur. J. Biochem., 1978, 86, 175 CAS.
- S. Ahmed, Ph.D. Thesis, Imperial College of Science, Technology and Medicine, London, 1991. D. W. Ribbons, thesis director..
- D. R. Boyd, N. D. Sharma, N. I. Bowers, J. Duffy, J. S. Harrison and H. Dalton, J. Chem. Soc., Perkin Trans. 1, 2000, 1345 RSC.
- V. Bui, T. V. Hansen, Y. Stenstrøm, D. W. Ribbons and T. Hudlicky, J. Chem. Soc., Perkin Trans. 1, 2000, 1669 RSC.
- A blocked mutant is a bacterial strain that lacks a particular enzyme in a metabolic pathway, resulting in accumulation of the metabolic intermediate produced immediately before the step blocked. In this case (Pp 39D), the enzyme that dehydrogenates cis-dihydrodiols to catechols is missing..
- All organisms used in this study were provided by David T. Gibson. (a) Pseudomonas putida 39D, see: D. T. Gibson, J. R. Koch and R. E. Kallio, J. Biol. Chem., 1968, 7, 2653 Search PubMed; (b) Escherichia coli JM109 (pDTG 601) expressing TDO, see: G. J. Zylstra and D. T. Gibson, J. Biol. Chem., 1989, 264, 14940 Search PubMed; (c) E. coli JM109 (pDTG 141) expressing NDO, see: M. J. Simon, T. D. Osslund, R. Saunders, B. D. Ensley, S. Suggs, A. Harcourt, W. C. Suen, D. L. Cruden and D. T. Gibson, Gene, 1993, 127, 31. Search PubMed.
- For E. coli JM109 (pDTG 602), see ref. 23b..
- It is difficult to introduce the catechol unit into highly functionalized compounds because of the harsh oxidizing conditions usually required. On the other hand, the presence of a catechol functionality in the starting materials alters the reactivity of the arene and may complicate the subsequent chemistry. Thus, the enzymatic introduction of this functionality into advanced intermediates has the potential to greatly simplify synthetic routes to catechol-containing targets..
- D. Gonzales, V. Schapiro, G. Seoane, T. Hudlicky and K. Abboud, J. Org. Chem., 1997, 62, 1194 CrossRef CAS; T. Hudlicky, K. Oppong, C. Duan, C. Stanton, M. G. Natchus and M. J. Laufersweiler, Bioorg. Med. Chem. Lett., 2001, in press.
- T. Hudlicky, E. E. Boros and C. H. Boros, Tetrahedron: Asymmetry, 1993, 4, 1365 CrossRef CAS.
- K. Königsberger and T. Hudlicky, Tetrahedron: Asymmetry, 1993, 4, 2469 CrossRef CAS.
- T. Hudlicky, E. E. Boros and C. H. Boros, Synlett., 1992, 391 CrossRef CAS.
- M. R. Stabile, T. Hudlicky, M. L. Meisels, G. Butora, A. G. Gunn, S. P. Fearnley, A. J. Thorpe and M. R. Ellis, Chirality, 1995, 7, 556 CrossRef CAS.
- L. A. Paquette, J. H. Kuo and J. Doyon, Tetrahedron, 1996, 52, 11625 CrossRef CAS.
- V. P. Bui, T. V. Hansen, Y. Stenstrøm and T. Hudlicky, Green Chem., 2000, 2, 263 RSC.
- Phenylcyclohexane was converted to the corresponding catechol with JM109 (pDTG602), see Scheme 3..
- T. Hudlicky, M. R. Stabile, D. T. Gibson and G. M. Whited, Org. Synth., 1999, 76, 77 CAS.
- G. Berti, B. Macchia, F. Macchia and L. Monti, J. Chem. Soc. C, 1971, 3371 RSC.
- W. E. Bachmann and L. B. Wick, J. Am. Chem. Soc., 1950, 72, 3388 CrossRef CAS.
- R. W. Murray, M. Singh, B. L. Williams and H. M. Moncrieff, J. Org. Chem., 1996, 61, 1830 CrossRef CAS.
- M. Tamura and J. Kochi, Synthesis, 1971, 303 CrossRef CAS.
- A. Garcia Martinez, A. Herrera, R. Martinez, E. Teso, A. Garcia, J. Osio, L. Pargada, R. Unanue, L. R. Subramian and M. Hanack, J. Heterocycl. Chem., 1988, 25, 1237 Search PubMed.
- J. K. Stille and B. L. Groh, J. Am. Chem. Soc., 1987, 109, 813 CrossRef CAS.
Footnote |
| † On leave from: Department of Chemistry and Biotechnology, the Agricultural University of Norway, N-1432 Ås, Norway. E-mail: E-mail: yngve.stenstrom@ikb.nlh.no. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |