Coordinated carbenes from electron-rich olefins on RuHCl(PPr3i)2†
Joseph N. Coalter IIIa, John C. Bollingera, John C. Huffmana, Ulrike Werner–Zwanzigera, Kenneth G. Caulton*a, Ernest R. Davidson*a, Hélène Gérardb, Eric Clotb and Odile Eisenstein*b
aDepartment of Chemistry and Molecular Structure Center, Indiana University, Bloomington, IN 47405-4001, USA. E-mail: caulton@indiana.edu; davidson@indiana.edu
bLaboratoire de Structure et de Dynamique des Systèmes Moléculaires et Solides (UMR 5636), Université de Montpellier 2, CC 14, Place E, Bataillon, 34095, Montpellier cedex 5, France. E-mail: odile.eisenstein@lsd.univ-montp2.fr
First published on UnassignedUnassigned4th January 2000
Abstract
Dehydrohalogenation
of RuH2Cl2L2
(L=PPr3i) gives (RuHClL2)2, shown to be a halide-bridged dimer by X-ray crystallography;
the fluoride analog is also a dimer. (RuHClL2)2 reacts with N2, pyridine and C2H4 (L′) to give RuHClL′L2, but with vinyl ether and vinyl amides, H2C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH(E) [E=OR, NRC(O)R′] such olefin binding is followed by isomerization to the heteroatom-substituted carbene complex L2HClRuC(CH3)(E). The reaction mechanism for
such rearrangement is established by DFT(B3PW91) computations, for C2H4 as olefin (where it is found to be endothermic),
and the structures of intermediates are calculated for H2CC(H)(OCH3) and for cyclic and acyclic amide-substituted
olefins. It is found, both experimentally and computationally, that the amide oxygen is bonded to Ru, with a calculated bond energy of approximately 9 kcal mol−1 for an acyclic model. Less electron-rich vinyl
amides or amines form η2-olefin complexes, but do not isomerize to carbene complexes. Calculated
ΔE values for selected ‘‘
competition’’ reactions reveal that donation by both Ru and the heteroatom-substituted X are necessary to make the carbene complex L2HClRuC(X)(CH3) more stable than the olefin complex L2HClRu(η2-H2CCHX).
This originates in part from a diminished endothermicity of the olefin→carbene transformation when the
sp2 carbon bears a π-donor substituent. The importance of a hydride on Ru in furnishing a mechanism for this isomerization
is discussed. The compositional characteristics of Schrock and Fischer carbenes are detailed, it is suggested that
reactivity will not be uniquely determined by these characteristics, and these new
carbenes RuHCl[C(X)CH3]L2
are contrasted to Schrock and Fischer carbenes.
CH(E) [E=OR, NRC(O)R′] such olefin binding is followed by isomerization to the heteroatom-substituted carbene complex L2HClRuC(CH3)(E). The reaction mechanism for
such rearrangement is established by DFT(B3PW91) computations, for C2H4 as olefin (where it is found to be endothermic),
and the structures of intermediates are calculated for H2CC(H)(OCH3) and for cyclic and acyclic amide-substituted
olefins. It is found, both experimentally and computationally, that the amide oxygen is bonded to Ru, with a calculated bond energy of approximately 9 kcal mol−1 for an acyclic model. Less electron-rich vinyl
amides or amines form η2-olefin complexes, but do not isomerize to carbene complexes. Calculated
ΔE values for selected ‘‘
competition’’ reactions reveal that donation by both Ru and the heteroatom-substituted X are necessary to make the carbene complex L2HClRuC(X)(CH3) more stable than the olefin complex L2HClRu(η2-H2CCHX).
This originates in part from a diminished endothermicity of the olefin→carbene transformation when the
sp2 carbon bears a π-donor substituent. The importance of a hydride on Ru in furnishing a mechanism for this isomerization
is discussed. The compositional characteristics of Schrock and Fischer carbenes are detailed, it is suggested that
reactivity will not be uniquely determined by these characteristics, and these new
carbenes RuHCl[C(X)CH3]L2
are contrasted to Schrock and Fischer carbenes.
We reported recently1 that 16-electron Ru(H)2Cl2L2 (L=PPr3i) can be dehydrohalogenated to give 14-electron RuHClL2, which has an unusual structure of a cis-divacant octahedron [eqn. (1), but vide infra].
 | (1) |
We have come to see that one special advantage of this geometry is that it has two empty sites cis and trans to H. Coordination of the substrate occurs preferably trans to Cl (smaller trans influence) and thus cis to H. This can facilitate the interaction between RuH and substrate. In contrast, the strong trans effect of hydride puts the empty orbital of five-coordinate d6 16-electron species of the MHX(CO)L2, etc., type trans to the hydride [eqn. (2)]
 | (2) |
and the resulting adduct is thus less suitable for rapid reaction between H and substrate S. This has major impact on the kinetics of substrate transformation.2
We report here a wide-ranging study of the surprising ways in which RuHClL2 rearranges olefins bearing π-donor substit uents on a vinylic carbon, as well as those carrying donor functionality at more remote sites. This furnishes an unusually simple preparation of a class of carbene ligands that does not rely on the conventional routes to carbenes: alkali metal alkyls and α-H abstraction [e.g., eqn. (3)], diazoalkanes [eqn. (4)], and addition of nucleophile, then electrophile to metal carbonyls [eqn. (5)]. This new method relies on the spontaneous (i.e., exothermic) rearrangement of free, then coordinated vinyl ethers to carbene complexes by a ruthenium monohydride [eqn. (6)]. In this paper, we investigate experimentally the scope of this reaction, as well as the origin of this thermodynamic preference using DFT calculations. The calculations will lead to a further clarification of these results in terms of the two ‘‘classes’’ of carbene ligands, the ‘‘Schrock type ’’ as formed in eqns. (3) and (4), and the ‘‘Fischer type’’, as produced in eqn. (5).
 | (3) |
| M+N2CHR→M(CHR)+N2 | (4) |
 | (5) |
 | (6) |
Results
(RuHClL2)2
This molecule is synthesized over a 12 h period in pentane [eqn. (1)]. The molecule shows diastereotopic Pri methyl hydrogens, which not only rules out a planar structure, but was interpreted earlier as indicating a ‘‘saw horse’’ monomeric structure A. Both the 1H and the 31P{1H} NMR spectra show only broadening, but no clear decoalescence at −95°C in toluene-d8. Crystals grown for an X-ray diffraction structure determination reveal the molecule to be the dimeric [RuH(μ-Cl)L2]2.3 The fluoride analog has also been shown to be a dimer by both multi-nuclear NMR and X-ray crystallography. However, since RuHClL2 forms an adduct (see Fig. 1 and Tables 1 and 2) with a reagent as weak as N2 (1 atm, 25°C, time of mixing), the inhibiting influence of the chloride bridges is insignificant, and the reagent will be written here as the monomer for simplicity. | ||
| Fig. 1 ORTEP drawing of the nonhydrogen atoms of RuHCl(N2)(PPr3i)2 showing selected atom labeling. The hydride was not located. | ||
| RuHCl(PPr3i)2(C6H9NO) | RuHCl(N2)(PPr3i)2 | |
|---|---|---|
| a R=Σ ‖Fo∣−∣Fc ‖/Σ∣ Fo∣; Rw=[Σw(∣ Fo∣− ∣Fc∣ )2/Σw∣ Fo∣2]1/2 where w=1/σ2(∣Fo∣). | ||
| Formula | C24H52ClNOP2Ru | C18H43ClN2P2Ru |
| Formula weight | 569.15 | 486.02 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/a | P21/c |
| a/Å | 16.124(5) | 7.9985(10) |
| b/Å | 11.289(4) | 8.9047(11) |
| c/Å | 16.313(5) | 16.614(2) |
| β/0 | 100.30(1) | 92.180(10) |
| U/Å3 | 2921.59 | 1182.5(2) |
| Z | 4 | 2 |
| T/°C | −171 | −171 |
| μ(MoKa)/cm−1 | 7.5 | 9.0 |
| Meas. reflections | 5136 | 6389 |
| Indep. reflections | 4692 | 6212 |
| Rint | 0.027 | 0.030 |
| Ra | 0.0460 | 0.0346 |
| Rwa | 0.0349 | 0.0675 |
RuHClL2 and equimolar pyridine form a 1:1 adduct in benzene within the time of mixing. The hydride chemical shift, −20.9 ppm, is sufficiently upfield to suggest that there is no ligand trans to hydride, and the coordinated pyridine shows five proton and five 13C chemical shifts, consistent with no facile rotation about the Ru←N bond. This is a symptom of a crowded environment, and addition of two equivalents of pyridine reveals formation of two new species, in addition to RuHClL2(py), which are assigned as bispyridine adduct RuHClL2(py)2 (a) and five-coordinate RuHClL(py)2 (b). The downfield hydride chemical shift of a (−12.9 ppm, t, 2JP-H=14 Hz) and the upfield signal with loss of coupling to one phosphine (free phosphine is also observed) for b (−20.0 ppm, d, 2JP-H=30 Hz) support these assignments and help illustrate this crowding.
The methyl protons of RuHCl(PPr3i)2 exchange over a period of several hours with the deuterons of C6D6. Presumably this takes place by generation of RuDCl(PPr3i)2 and C6D5H, followed by the ruthenium deuteride scrambling into the phosphines as discussed below. The phenomenon is detected by 1H NMR via a large increase in the protio signal of benzene-d6, coupled with a decrease and broadening of the signals for Ru–H and the Pri methyl groups when RuHCl(PPr3i)2 is placed in a flame-sealed NMR tube and periodically monitored for 2 days.
Reactivity of RuHClL2 towards hydrocarbon olefins
RuHClL2 binds ethylene (1 atm or equimolar, 20°C) to give a 1:1 adduct. The hydride chemical shift, −22.0 ppm, is still sufficiently upfield to suggest that there is no ligand trans to hydride, consistent with structure B.
Although this is the ground state structure of this molecule, reaction of RuHClL2 with excess C2D4 in C6H6 for 1 h at 20°C shows (2H NMR) deuteration of the H on Ru and also the methyl groups of coordinated PPr3i. This is indicative of reversible insertion of C2D4 into the Ru–H bond and it also indicates that the 16-electron olefin hydride form (B) is more stable than Ru(C2H5)ClL2. Deuteration of the Pri methyls is accounted for by reversion to RuDCl[P(CH(CH3)2)3]2 and then CH3/RuD scrambling, as was independently established for this species deuterated by two independent methods. Insertion of ethylene into Ru–H is also evidenced by the formation of ethane, detected by 1H NMR. Its formation most likely occurs by alkane elimination from unstable Ru(C2H5)ClL2, which has oxidatively added an Pri methyl group (C–H). This C–H activation also offers an additional explanation for deuteration of the Pri methyl with C2D4. The metal-containing products after CH3CH3 elimination could not be identified. There is no isomerization of ethylene into the carbene ligand CH(CH3).
RuHClL2 also reacts within 30 min with the olefins 1-hexene and styrene, but only 10% adduct is formed, with unreacted RuHClL2 comprising the bulk of the resulting mixture at 25°C. Both these adducts are identified by a new hydride signal in 1H NMR (−23.7 ppm, m, for 1-hexene and −22.1 ppm, apparent triplet, for styrene) and corresponding 31P{1H} NMR AB patterns centered at 37.2 ppm (2JP-P=287 Hz) for 1-hexene and at 85.9 ppm (2JP-P=34 Hz) for styrene. The spectroscopic similarity of the ethylene, 1-hexene, and ethyl vinyl ether adducts and large difference of the styrene adduct suggests that styrene may coordinate differently (perhaps as η2: η1–2-vinyl:arene or η2–4-arene). Longer reaction times yield no carbenes, but only complex mixtures of products.
Reactivity of RuHClL2 towards vinyl ethers
RuHClL2 rapidly effects a formal 1,2-hydrogen migration [eqn. (7)] of vinyl ethers into the coordinated carbene. The reaction occurs for a variety of groups R, including those with SiMe3, ether, alcohol, tertiary amino, fluoro, and epoxide functionality. All products show diastereotopic Pri methyl groups, consistent with the presence of three different substituents, H, Cl and carbene, on Ru. While this reveals nothing about the square pyramidal vs. trigonal bipyramidal geometry around Ru, the hydride chemical shifts (Table 3), rather far upfield (−21 ppm), could be interpreted in terms of the hydride being approximately trans to an empty site (i.e., square pyramidal). The hydride chemical shift is thus also perhaps the most generally sensitive indicator of whether there is a ligand (from functionality in the substituent R) coordinated trans to hydride. The similarity of the hydride chemical shift for R=Et (Table 3) to those for all components with functionalized alkyl groups in eqn. (7) suggests that none of the latter O, N or F donors coordinates to Ru.4 Perhaps this is caused by the difficulty of forming six-membered rings. For the case of F, this conclusion is reinforced by a 19F chemical shift that lies within 1 ppm of that of the free olefin, and by the absence of coupling to F in the 31P NMR spectrum. Even at −80°C, 19F NMR spectra reveal that F remains uncoordinated.| δ Ru–H | 2JP-H/Hz | δ RuC | 2JP-C/Hz | δ Ru–P | |
|---|---|---|---|---|---|
| a 1H data reported in CD2Cl2.b Not measured.c Not resolved (broad).d Cyclic.e Dimetal carbene.f All NMR data in CD2Cl2. | |||||
| Vinyl ethers | |||||
| OEt | −21.7 | 23 | 290 | 9.7 | 58.2 |
| OCH2CH2CH2–ad | −18.2 | 22 | 287 | 9.4 | 60.1 |
| OCy | −19.4 | 23 | b | b | 57.4 |
| OSiMe3 | −18.0 | 22 | 284 | 8.4 | 58.6 |
| OCH2CH2OBun | −21.3 | 22 | 289 | 9.5 | 57.8 |
| CH2CH2OH | −21.1 | 22 | 289 | 8.4 | 56.4 |
| (OCH2CH2)2OH | −21.4 | 21 | b | b | 56.3 |
| –OCH2CH2O–e | −21.2 | 22 | 289 | 9.1 | 57.9 |
| OCH2CH2F | −21.3 | 22 | b | b | 57.8 |
| OCH2CH2NEt2 | −21.4 | c | b | b | 56.4 |
| OCH2CHOCH2 | −21.4 | 22 | b | b | 57.7 |
| Vinyl amides | |||||
| N(Me)C(O)Mef | −17.2 | 26 | 265 | 9.7 | 50.4 |
| NC(O)CH2CH2CH2–df | −19.6 | 26 | 262 | 10 | 49.3 |
 | (7) |
When O is from an epoxide ring, the presence of a chiral carbon β to the vinyloxy oxygen necessitates phosphine inequivalence. From the magnitude of their 31P{1H} NMR (Fig. 2) chemical shift difference [Δδ=only 0.09 ppm (15 Hz), 2JP-P=220 Hz], it is safe to assume that the chiral center is far from the phosphorus atoms, indicating no epoxide binding to ruthenium.
2, showing the inequivalence of the phosphorus nuclei caused by the epoxide carbon asymmetry. Starred peaks are impurities and the arrows indicate the outer lines of the AB pattern.](/image/article/2000/NJ/a907624g/a907624g-f2.gif) | ||
| Fig. 2 31P{1H} NMR (162 MHz) spectrum of RuHCl[C(Me)(OCH2CHOCH2)](PPr3i)2, showing the inequivalence of the phosphorus nuclei caused by the epoxide carbon asymmetry. Starred peaks are impurities and the arrows indicate the outer lines of the AB pattern. | ||
The case of dihydrofuran [eqn. (8)] is interesting as it shows that a cyclic internal vinyl ether is also easily isomerized.5 The hydride chemical shift is consistent with no donation to Ru by the ether β-oxygen, just as it does not occur for the products in eqn. (7). Using the symmetrical difunctional vinyl ether as in eqn. (9), with slow addition of the vinyl ether to RuHClL2, a dimetal carbene is produced.
 | (8) |
 | (9) |
The presence of inequivalent (i.e., H and Cl) substituents on
Ru, together with the carbene plane not
eclipsing the RuP2
plane,6
makes two isomers (C and D) possible for an unsymmetrically substituted carbene, CRR′. Indeed, upon lowering the temperature in toluene-d8, one observes broadening, then decoalescence and sharpening of the Ru–H, the C–CH3 and the OCH21H NMR signals of two isomers (C and D) with a population ratio of more than 10:1. The 31P{1H} NMR confirms this,
with decoalescence at about −50°C and two separate resonances
(major isomer at 55.8 and minor at 51.2 ppm) resolved below −70°C. Lineshape analysis of the 31P{1H} NMR spectrum
gives ΔG‡=9.8 kcal mol−1 for C→D at −60°C. The assignment of isomer structure to spectra was done by NOE
experiments of the hydrides at −95°C. The large (5 ppm) difference in the hydride chemical shift of these isomers
supports their being isomeric around the RuC bond and not around the C(carbene)–OEt bond. These observations are significant
because (1) no previously known compound has had suitable symmetry to prove that carbene rotation is rapid at 25°C and
(2) carbene rotation is an essential step in the olefin metathesis6
process (i.e., forming the metallacyclobutane).

 | ||
| Fig. 3 31P{1H}
NMR monitoring of reaction progress when RuHClL2 and H2CCH(OEt) are combined at −65°C in toluene-d8
. Doublet in RuHClL2 signal is due to incomplete hydride decoupling. Arrow shows initial growth of carbene product at 0°C. | ||
Carbene product formation demands cleavage of the C–D bond in H2CC(D)(OEt), but at least three mechanisms (Scheme 1) can be envisioned: (a) direct C–D oxidative addition
to the ruthenium hydride, followed by hydrogen migration
to the vinyl Cβ, (b) primary addition of Ru–H to CC, followed
by α-hydrogen migration to Ru, (c) concerted 1,2-hydrogen migration
within the coordinated olefin. These have distinct predictable consequences for the deuterium label since (a) mixes RuH
with C–D, (b) migrates D exclusively to Ru only after the
Ru–H has been cleaved, and (c) migrates D exclusively to
the CH2
carbon. In fact, when RuHClL2 and H2CCD(OEt) are
combined at −40°C and observed by 2H NMR beginning at −20°C,
one sees immediately RuDCl(olefin)L2 in which
there is also D in the phosphine methyl groups, and some
free HDCCH(OEt). These indicate reversible olefin binding to
Ru and reversible migration of H (or D) from Ru to both olefinic
carbons [eqn. 10)].
 | ||
| Scheme 1 | ||
The scrambling of D into the phosphine methyls was already
established as a characteristic of RuDClL2 itself. It is thus
clear that Ru–H adds in both directions to the olefin, but only one of these leads to carbene product; the regiochemistry of carbene
production is not caused by selectivity in the initial H migration step. By 0°C, carbene product grows in, with D both at Ru (25%) and at the carbene methyl (75%). Thus, attempts to establish mechanism by quantitative comparison to the
predictions of eqn. (10) are frustrated by general isotope scrambling.8 However, the significantly slower reaction
rate observed using H2CCD(OEt) establishes that C–D cleavage occurs before or at the rate determining step. From the scrambling patterns observed, we propose that the most likely mechanism of carbene formation combines mechanism b of Scheme 1 and eqn.
(10).
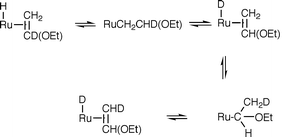 | (10) |
Reactivity of RuHClL2 towards vinyl amides
The vinyl amides are also transformed into the carbenes shown in eqn. (11). The chemical shifts of the carbene carbon, of the hydride, and of 31P are all sufficiently different from those in eqn. (7) to suggest the possibility of amide oxygen coordination. Interestingly, there is a large difference in the 1H NMR chemical shift of Ru–H and in the ν(CO) of the pendant amide between the products from cyclic and acyclic vinyl amides (R,R′=Me: δ=−17.2 ν(CO)=1599 cm−1; R,R′=CH2CH2CH2: δ=−19.6 ν(CO)=1640 cm−1). These data show that while both compounds may have CO metal-bound, the geometric constraints at the cyclic species hinder its binding relative to the carbene derived from acyclic N-methyl-N-vinyl acetamide. Carbonyl
oxygen coordination was confirmed by an X-ray crystal structure determination for the product from 2-vinyl pyrrolidinone (Table 4 and Fig. 4). The structure is derived from the five-coordinate carbene complexes Ru(carbene)Cl(H)L2 by addition of the pendant carbonyl functionality of the carbene in the site trans to hydride. The inner coordination sphere has idealized mirror symmetry, which extends even to the conformations of all Pri groups. Bond angles in the six-coordinate
polyhedron are generally within 10° of the ideal 90°, except
for within the five-membered ring of the bidentate carbene/amide ligand [79.45(17)°]; also the phosphorus nuclei
deviate visibly from octahedral symmetry [∠P–Ru–P=158.67(5)°] and they
bend main ly towards hydride and sligtly towards chloride. The Ru–C(carbene) distance is short (but not as short as to a carbonyl ligand9) and the distance to Cl trans to carbene is unusually long [2.5387(14)Å]. The Ru←O distance is not
especially long, in spite of its being trans to hydride.
There is no trace of interaction between the mutually cis
ligands hydride and carbene; hydride lies in the nodal plane of the RuC bond. The distance from N28 to the carbene carbon is
8σ (0.063 Å) longer than to the carbonyl carbon (C24), which suggests that the N–C π bond is more in the amide group, and less to the carbene carbon [eqn (12)]. | (11) |
| Ru(1)–Cl(2) | 2.5387(14) | O(23)–C(24) | 1.245(6) |
| Ru(1)–P(3) | 2.3658(15) | N(28)–C(24) | 1.350(6) |
| Ru(1)–P(13) | 2.3664(15) | N(28)–C(27) | 1.476(6) |
| Ru(1)–O(23) | 2.273(3) | N(28)–C(29) | 1.413(6) |
| Ru(1)–C(29) | 1.880(5) | Ru(1)–H(1) | 1.25(4) |
| P(3)–Ru(1)–P(13) | 158.67(5) | C(24)–N(28)–C(29) | 118.0(4) |
| P(3)–Ru(1)–O(23) | 99.96(10) | C(27)–N(28)–C(29) | 129.8(4) |
| P(3)–Ru(1)–C(29) | 92.84(14) | Ru(1)–C(29)–N(28) | 114.7(3) |
| P(13)–Ru(1)–O(23) | 100.57(10) | Ru(1)–C(29)–C(30) | 133.8(4) |
| P(13)–Ru(1)–C(29) | 96.51(14) | N(28)–C(29)–C(30) | 111.5(4) |
| O(23)–Ru(1)–C(29) | 79.45(17) | Cl(2)–Ru(1)–H(1) | 96.4(16) |
| Ru(1)–P(3)–C(4) | 116.40(19) | P(3)–Ru(1)–H(1) | 78.3(16) |
| Ru(1)–P(3)–C(7) | 114.40(7) | P(13)–Ru(1)–H(1) | 82.1(16) |
| Ru(1)–P(3)–C(10) | 112.53(18) | O(23)–Ru(1)–H(1) | 171.9(16) |
| Ru(1)–P(13)–C(14) | 113.92(18) | C(29)–Ru(1)–H(1) | 92.7(16) |
| Ru(1)–P(13)–C(17) | 115.80(19) | Cl(2)–Ru(1)–P(3) | 88.06(4) |
| Ru(1)–P(13)–C(20) | 113.68(20) | Cl(2)–Ru(1)–P(13) | 85.71(5) |
| Ru(1)–O(23)–C(24) | 106.2(3) | Cl(2)–Ru(1)–O(23) | 91.47(9) |
| C(24)–N(28)–C(27) | 112.2(4) | Cl(2)–Ru(1)–C(29) | 170.90(15) |
2, showing selected atom labeling.](/image/article/2000/NJ/a907624g/a907624g-f4.gif) | ||
| Fig. 4 ORTEP drawing of the nonhydrogen atoms of RuHCl[C(Me)NC(O)CH2CH2CH2](PPr3i)2, showing selected atom labeling. | ||
 | (12) |

The amide reactions proceed considerably slower than those with vinyl ethers, taking 2–4 days to react completely. After 1 h in C6D6, the mixture of 2-vinyl pyrrolidinone and RuHClL2 exhibits 1H and 31P NMR signals for three major intermediates, as well as for starting material and final product. One intermediate exhibits an AX pattern (δ 57.6, 33.4; 2JP-P=274 Hz) in the 31P spectrum and an apparent triplet in 1H NMR (δ −29.2, 2JP-H=34.8 Hz) while the other two show singlets in 31P (δ 85.6, 74.7) and Ru–H doublets in 1H (δ −10.1, −19.6; 2JP-H=33.0, 35.1 Hz, respectively). These latter intermediates are consistent with dissociation of phosphine and indeed free phosphine is observed by 31P NMR. Upon completion of the reaction, phosphine re-coordination occurs, with the only product seen being the amido-substituted carbene. Although the exact nature of all intermediates cannot be inferred (coordination by olefin, N, η2-carbonyl, or a combination of these groups all produce inequivalent phosphines), the multiple coordination modes of vinyl amides, coupled with the possibility of required phosphine dissociation, help explain the kinetic sluggishness of this reaction.
Reaction of equimolar RuHClL2 and N-vinyl phthalimide leads to E, which does not rearrange to a carbene isomer over 20 h at 25°C. After this time, substantial decomposition of product occurs in solution. Complex E shows inequivalent phosphines, with 31P chemical shifts of 35.4 and 57.9 ppm and a JP-P′ of 278 Hz. These values are very close to those of the bis phosphine intermediate detected in reaction with 2-vinyl pyrrolidinone. The large difference in chemical shift of the carbonyl groups by 13C{1H} NMR (10 ppm), coupled with a hydride chemical shift similar to the carbenes from vinyl amides, lends evidence for oxygen coordination. Thus, while one keto group on N permits isomerization to a carbene complex, two keto groups apparently leave the nitrogen too weak a donor to stabilize a carbene.
It is of interest that 9-vinylcarbazole (F) is not too bulky to bind to RuHClL2, but the reaction does not proceed to a carbene complex, perhaps because the nitrogen lone pair is too involved in the arene π system to stabilize the electrophilic carbene G, which would also be very crowded. It is clear that nitrogen is not the donor site in the observed adduct since the phosphines are inequivalent, as they would be in a π-olefin structure.

Reactivity with a conjugated diene ether substrate
Addition of 10 equiv. of the conjugated diene, 3-methylene- 2,3-dihydrofuran, to a solution of RuHCl(PPr3i)2 in C6D6 results in complete isomerization to 3-methylfuran within 10 min. [eqn. (13)]. | (13) |
No reaction occurs from 3-methylfuran with RuHCl(PPr3i)2 at room temperature over several hours. This isomerization is consistent with addition of Ru–H across the terminal alkene to give the tertiary alkyl, followed by β-H elimination to achieve aromatic stabilization in the resulting furan.
DFT computation of the ethylene→methyl carbene isomerization
The isomerization of free ethylene into free methyl carbene is known to be a very endothermic reaction (80 kcal mol−1). Recent laser flash photolysis experiments and ab initio calculations on propene have shown that the energy of reaction is 60 kcal mol−1 endothermic.10 Isomerization of free olefin is thus unlikely to happen in less extreme conditions (i.e., in solution).The reaction path for transforming ethylene into its isomer C(H)CH3 in the presence of RuHCl(PH3)2 [Ru] (as a model for RuHClL2) was determined with DFT(B3PW91) calculations (Fig. 5). As mentioned earlier, the 14-electron fragment RuHClL2 does not exist as an isolated species. However, it is a reasonable assumption to consider that the olefin causes the dissociation of the dimer into two monomeric RuHClL2, which is stabilized through coordination to the olefin. For the sake of discussion, we will consider that the starting entities are thus [Ru] and C2H4.
 | ||
| Fig. 5 DFT(B3PW91) optimized structures and energies for RuC2H5Cl(PH3)2 isomers. | ||
The olefin coordinates [Ru]
trans to Cl to form a 16-electron intermediate 1, which is a square-based pyramid with hydride at the apical site. This adduct has the classical geometry of numerous five-coordinate d6 species. The olefin is coordinated opposite to the ligand with the smaller trans
influence (Cl). This combination of ligands on the metal forces the olefin to be cis to the hydride, which is favorable for further reaction between these two ligands. The orientation of the olefin also is a favorable factor since the CC bond is found to be aligned with Ru–H. This preferred orientation minimizes the steric effect between the phosphine ligands and the olefin
and maximizes the stabilizing cis interaction between the hydride and the olefin,11
although it should be noted that
the cis effect alone does not make the olefin tilt toward H. The olefin adduct has no other remarkable structural aspects. The binding energy of [Ru] to C2H4 is quite large (39.2 kcal
mol−1), which characterizes the high unsaturation of [Ru].
The insertion of the olefin into the Ru–H bond is a facile process since the transition state is found to be only 7.6 kcal mol−1 above 1. It produces an ethyl complex, 2, which is less stable than 1 by 6.2 kcal mol−1. This ethyl complex has a remarkably strong β agostic C–H bond since the Cβ–H bond length is equal to 1.221 Å, while Ru–H is significantly short, 1.774 Å. At
the same time, the C–C bond length is significantly shorter (1.482 Å) than that of a single C–C bond and the two Ru–C distances
do not differ greatly (2.070 Å and 2.293 Å), although the
initial difference in bond lengths between Ru–Cα and Ru–Cβ has been increased. The transition state TS1-2 between 1 and 2 has the expected features of an unsaturated ligand inserting
into an Ru–H bond: the ethylene is more unsymmetrically bonded
(Ru–Cα=2.107,
Ru–Cβ=2.215 Å) than in 1 and less than in 2. However, in a nonintuitive manner, Cβ has moved closer to Ru than it was in 1, in an apparent attempt to assist in H–Cβ bonding. The C–C bond length (1.444 Å) is intermediate between that in 1 and in 2. The main change in structure is thus the displacement of H as measured by the Cl–Ru–H angle (97.6°, 144.0°, 159.0° as one goes from reactant to product). According to
the Hammond postulate, TS1-2 should resemble more 2, to which it is closer in energy, and this is true for the Cl–Ru–H angle
and the Ru–C(α distance. However, the behavior of Ru-Cβ illustrates some limitation in using the Hammond postulate.
With regard to forming the HRuC(H)Me moiety, the geometry of 2 is poorly adapted for migrating a hydrogen from
Cα to the metal. Rotation of the ethyl group is thus necessary to put an α hydrogen in proximity to the nearest empty orbital of the metal. Rotation of the ethyl is energetically demanding (12.1 kcal mol−1) since it is necessary to break the unusually strong Cβ–H agostic bond in addition to rotating about the single Ru–Cα bond. The transition state TS2-3
has no agostic interaction (closest Ru···H distance= 2.6 Å) and thus the C–C bond length is typical of that of a single bond (1.523 Å). The rotation of the ethyl group leads to 3, where the ethyl group has an α agostic hydrogen. The energy of this intermediate is 9.8 kcal mol−1 above 2. This is in great part due to the large difference in the strength of the agostic interactions. In 3, Cα–H
(1.13 Å) is much less elongated than Cβ–H in 2 and, consequently, the agostic H is much further away from Ru (2.334 Å in 3). In addition, the C–C bond (1.517 Å) is close to that of a single C–C bond.
Since rotation about the Ru–Cα bond is energetically demanding, we searched for an alternative path. Inversion at the metal center would achieve the requirement to put an α hydrogen in proximity to an empty metal orbital. The transition state, TS′2-3, for this transformation was located 33.1 kcal mol−1 above 2. The geometry of this TS is square planar. Its high energy shows the strong energetic preference for a bent geometry at a d6 four-coordinate center.
The α migration from 3, to the final hydrido carbene complex 4, is an easy process (ΔE‡=6.4 kcal mol−1). Remarkably, 3 and 4, although they are very different in bonding and in formal valence electron count, are isoenergetic. Whatever determines the energies of both of these is responsible for [Ru] being unable to isomerize ethylene to ethylidene: the olefin form 1 is more stable and even the β agostic ethyl, 2, which the calculations suggest to be kinetically accessible at 25°C, is significantly more stable than 3 and 4. This accounts for the deuterium scrambling and the absence of an observable quantity of ethylidene complex in the reaction of C2D4.
The final product 4 has the geometry of a trigonal bipyramid distorted towards a Y shape through large Cl–Ru–C (129.5°) and Cl–Ru–H (142.5°) angles. The plane of the carbene eclipses the P–Ru–P axes. This conformation permits the empty carbene p orbital to interact with the highest occupied d Ru orbital (H). Therefore, bulky phosphines could force a rotation of the carbene plane. It has been shown that the conformation of the carbene in the related RuCl2(PR3)2(CHR) depends on the nature of the substituents at the phosphine and carbene.6 The transition state TS3-4 to form 4 has already many geometrical aspects of the final product despite being energetically equidistant from 3 and 4. The carbene complex is fully formed since the carbon bonded to the metal is planar (sum of the angles at C=360°). The Ru–C distance is equal to 1.879 Å, very close to that in the final product (1.855 Å). The Ru–H bond is also almost fully formed (1.670 Å) vs. 1.606 Å in 4. The major difference between TS3-4 and 4 lies in the angles between the three ligands. The Cl–Ru–C angle varies from 145.0° (TS3-4) to 129.5° (4), while ∠H–Ru–C varies from 53.5 to 87.9°, accordingly. In this last step of the reaction path, as well as in the step of the insertion of ethylene into the Ru–H, there is no synchronous variation of all the coordinates from reactants to products (in contrast to the predictions of the Hammond postulate).
DFT computation of the vinyl ether→alkoxy carbene isomerization
The model chosen for CH2C(H)OR is CH2C(H)OCH3. The geometry of the vinyl ether coordinated to [Ru] has been optimized (Fig.
6). The overall geometry is that of a square pyramid with an apical hydride ligand. The olefin prefers to coordinate the metal in the mirror plane of the molecule, as does ethylene. Even in the absence of large phosphines, the conformation observed experimentally is thus shown to be preferred. Both isomers 5 and 6 (Fig. 6) are found to be minima on the potential energy surface with a preference of 0.9 kcal mol−1
for 6. This almost negligible difference in energy is not due to an O···Ru interaction in 6 since the oxygen is outside of the bonding range to the metal (Ru···O distance〉3 Å). | ||
| Fig. 6 DFT(B3PW91) optimized structures for isomeric RuC2H4(OCH3)Cl(PH3)2 isomers. | ||
Comparing the ethylene and the CH2C(H)OCH3 adducts, it appears
that the substitution by OCH3 has no significant effect
on the geometry of the complex. In 6, there is a slight decrease
in the non-bonding distance between the unsubstituted carbon of CH2C(H)OMe and Ru with respect to the
corresponding
distance in the case of an ethylene ligand (2.19 Å
vs. 2.23 Å),
consistent with the slight attraction described above. Remarkably,
the Ru–olefin bond dissociation energy (BDE≈39 kcal mol−1)
is also not affected by the presence of OCH3
(average difference
of 3.0 kcal mol−1 with the greater dissociation energy
for ethylene). While OCH3 certainly increases the electron-donating
ability of the π system, it decreases its π-accepting capability. These two effects act in opposite directions to maintain the binding energy to the metal constant. This emphasizes that [Ru], although strongly electron-deficient, is not acting
solely as a Lewis acid but certainly has important back-donating
capabilities.
The structures of several conformations (all minima) of the
[Ru]C(CH3)(OCH3) isomer are also shown in Fig. 6. In the two most stable conformations, the carbene is in the mirror plane of the molecule. These two conformations, 7 (OCH3cis to H with respect to the RuC bond) and 8 (OCH3trans to H with respect to the RuC bond), are only 0.25 kcal mol−1 apart and thus at
the same energy. The conformation 9 with the carbene plane
perpendicular to the mirror plane is calculated to be 4.1 kcal
mol−1 above the most stable conformation. Since 9
would
be highly disfavored on steric grounds in the real system, the
two preferred conformations are 7 and 8. Finally, a carbene
complex in which the oxygen would coordinate Ru (I) was sought
as a minimum on the potential energy surface without success.
Any such structure optimized to 8. No 18-electron C-
and
O-bonded carbene complex is more stable than the 16-e unsaturated
η1-bonded C(CH3)(OCH3) species.

The preferred orientation C(CH3)(OCH3) is thus rotated by 90° with respect to that in 4, the C(H)CH3 complex. The coordination around the metal is also different for the two carbene ligands. For C(H)CH3, ∠Cl–Ru–C is equal to 129.5° and ∠Cl–Ru–H is equal to 142.5°. With the OCH3 substituent ∠Cl–Ru–C is 161.6° (7) and 167.5° (8), while ∠Cl–Ru–H is 107.9° or 107.8°, respectively. The coordination at the metal has thus changed from a Y shape [C(H)Me] to a T shape [C(CH3)OCH3]. It should be noticed that the change from Y to T shapes at Ru is associated with the rotation of the carbene and not solely with the presence of the OMe substituent. Thus, in 9 where the C(Me)(OCH3) is perpendicular to the mirror plane, the coordination geometry at Ru is similar to that of 4. The preference of different carbene conformations for the Y and T shape complexes has its origin in the back-bonding from Ru to the carbene. In the Y shape, as mentioned above, the highest occupied d orbital is dx2 −y2 resulting in a carbene perpendicular to the mirror plane. In the T structure, the dxy and dxz (J) orbitals are both destabilized by a Cl lone pair and are thus each good candidates for back-donating into carbene. An additional increase in back-bonding is caused by bending the phosphine ligands away from the carbene. This is only possible when the carbene lies perpendicular to the Ru–P bonds as in 7 or 8 so that its π orbitals can accept electrons from dxz.

The OCH3 group acts as a π donor in the carbene and not in the olefin adduct as shown by the significantly shorter C(sp2)–O bond in the former systems. The π donation of the OCH3 group weakens the electron donation from Ru–C as proven by the comparison of 4 and 6 or 7 and 8, where the Ru–Cα bond is the shortest for C(H)CH3.
The presence of the π-donor group OCH3
has also significantly decreased the difference in energy between the olefinic adduct and its carbene isomers (Fig. 7). In the case of ethylene, the isomerization was endothermic by 15.9 kcal mol−1. In the case of vinyl ether, the isomerization is calculated to be essentially
thermoneutral. The isomer of the carbene complex with the OMe group on Cβ (9′) has been calculated to be 21.1 kcal mol−1 above the most stable olefin adduct. The difference in energy between the olefin adduct CH2
CHX and the carbene isomer increases in the order X=OMe on Cα (0), H (15.9) OMe on Cβ (21.1 kcal mol−1). Thus, the OMe group greatly facilitates the formation of the isomer when positioned on Cα, but significantly disfavored the formation of carbene when on Cβ. Despite the fact
that the initial structure employed had O in the vicinity of Ru, the optimized geometry of structure 9′ shows that O has moved away from the metal. The carbene orientation and the coordination at Ru is similar to that of 9. Despite the presence of unsaturation at Ru, the oxygen coordination does not bring any stabilization. In fact, angles Ru–Cα–Cβ (132°) and Cα–Cβ–O (115°) in 9′ open to keep O from being too close to the phosphine ligand. Thus, while coordination of olefin does not show any significant preference for having OMe at a given position (structure 5
and 6), only 6 would lead to the carbene
product.
 | ||
| Fig. 7 Comparison of the influence of substituent X on isomer stabilities. | ||
It is especially illuminating to relate the difference in energy between the unsubstituted or substituted olefin adduct and the carbene complex to the corresponding values in the absence of [Ru]. Thus, both the Ru fragment [eqn. (14)vs. (16)] and the OCH3 substituent [eqn. (14)vs. (15)] drastically decrease the isomerization energy of olefin into carbene since both can donate to the empty p orbital of the carbene. The competition between [Ru] and OCH3 in donation to the same empty carbene p orbital results in some interesting features in the following isodesmic reactions.
 | (14) |
 | (15) |
 | (16) |
 | (17) |
 | (18) |
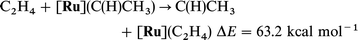 | (19) |
Eqn. (18) and (19) show that OCH3 and [Ru] independently stabilize
the carbene isomeric form with respect to the olefinic form by large amounts. However, eqn. (20)
[in comparison to eqn. (19)] shows that [Ru] is less efficient in stabilizing
methoxycarbene than methyl carbene. This difference could be due to
the influence of OCH3 on the olefin and/or the carbene. However, there is no significant difference in BDE to [Ru] for
ethylene and CH2CHOCH3 as shown by eqn. (21). Thus, [Ru] stabilizes the non-substituted carbene significantly better [eqn. (22)]
than the C(CH3)(OCH3). While OCH3 diminishes the stabilizing influence of [Ru] to the carbene due to the fact that both are in competition to donate to the same empty orbital of the
carbene, it should be emphasized that the cooperative effect of both OCH3 and Ru is indispensable to compensate the large
difference (79.1 kcal mol−1) in energy between the olefin and
its isomeric carbene form [eqn. (14)].
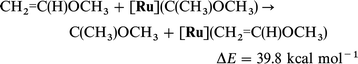 | (20) |
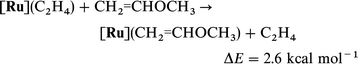 | (21) |
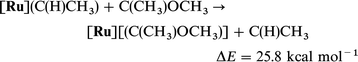 | (22) |
DFT computational study of the amido carbene complex
The amido carbene complex in which PPr3i was replaced by PH3 (10) was optimized with no symmetry restriction. The optimized structure is in good accord with the experimental structure (Fig. 8). All metal-to-ligand distances are close to the experimental values with the exception of Ru···O, which is 0.1 Å too long. However, the calculated Ru···O is sufficiently short (2.370 Å) to clearly indicate the presence of a Ru···O bond. The calculated geometry within the Ru–C–N–C–O moiety mimics closely the experimental values. The N center, which is calculated to be planar, is closer to the C of carbonyl (N–C=1.382 Å) than to Cα (1.391 Å). The five-membered ring has the usual envelope shape. | ||
| Fig. 8 DFT(B3PW91) optimized structures for RuHCl(PH3)2 with amido olefins. | ||
The binding energy of oxygen to the metal can be estimated from the differences in energy between 10 and optimized structures with no Ru···O interaction. To save computational time, a simplified system 11 in which the five-membered pyrrole ring is truncated, has been selected. The optimized structure of 11 (Fig. 8) gives results very similar to that of 10. This model will thus be used for further study of the system.
The structure 12, lacking Ru···O interaction and resulting from a rotation about the Cα–N bond, was located as a minimum (Fig. 8). The energy of 12 is 8.6 kcal mol−1 higher than 11. Another minimum 13 (not shown), obtained from 12 by 180° rotation about the Ru–Cα bond, was located and is calculated to be 9.8 kcal mol−1 above 11. The Ru···O bond dissociation energy is thus estimated to be about 9 kcal mol−1.
There are some interesting geometry changes in going from 11 to 12. In losing the Ru···O interaction, the carbene rotates by 43° (13 has similar geometrical features). This unusual conformation of the carbene is thus intermediate between that of C(H)CH3 and C(CH3)(OCH3). There is an apparent corre lation between the donating power of the substituent on the carbene and the conformation of the carbene in the complex. We have seen that the orientation of the carbene was closely connected to the coordination geometry at the metal [Y for CHMe and T for C(OMe)(Me)]. In the case of 11 (∠Cl–Ru–Cα=162.0° and ∠Cl–Ru–H=114.5°), the metal coordination is also intermediate between the C(H)CH3 and C(CH3)OCH3 cases. In 12, the nitrogen (still planar) is closer to Cα (1.380 Å) than to the C(O) carbon (1.406 Å) in contrast to 11. The π donation of the nitrogen is thus more important towards the carbene in 12 than in 11, due to the fact that the electron accepting ability of the acyl group is no longer enhanced by coordination to Ru.
Two olefin adducts, 14 and 15, with a NH(CO)(CH3) substituent have been optimized. Isomer 14 is 5.5 kcal mol−1 more stable than 15
and has the amide oxygen coordinated to Ru with only a minor decrease in the P–Ru–P angle (160.7° in 14 and
166.6° in 15). Thus, the more bulky phosphine used experimentally should not prevent the coordination. The proposal that O is coordinated to the metal in the olefin adduct 14 is
supported by the structure of the olefin adduct of CH2
CH(phthalimide).
The difference in energy between the most stable olefin complex 14 and the more stable carbene isomer 11 is calculated to be 3.1 kcal mol−1 in favor of the olefin adduct (Fig. 7). This small difference in energy, together with inclusion of steric effects (which favor the carbene over olefin complex), accounts for the fact that isomerization of the olefin substituted by the amido group has been observed. In this case, two factors contribute to make the carbene isomer energetically accessible: π donation of the nitrogen amide and coordination of the amide oxygen to Ru, which in turn probably enhances the electron-donating ability of Ru toward the carbene. It is, however, probable that π donation of the nitrogen lone pair is a major component since no isomerization is observed in the case of vinyl phthalimide.
Discussion
The multistep reaction path that is described here is analogous to that of the isomerization of terminal alkyne into vinylidene in the presence of Ru.12 However, while the path is exothermic from the acetylene adduct to the vinylidene complex [eqn. (23)], it is endothermic in the case of ethylene [eqn. (24)]. At the heart of this is the difference in energy between the isomeric species [ΔE(C2H2−CCH2)=40
kcal mol−1]13 and [ΔE(C2H4−C(H)CH3)=79.1 kcal mol−1]. This large difference, which is due to the fact that the third bond of a triple bond is much weaker than the π bond of a double bond, controls the thermochemistry of the reaction even in
the presence of the metal. The presence of a π-donor group like OCH3
lowers the endothermicity of the isomerization of free olefin to 50 kcal mol−1, thus approaching that of free alkyne. Lowering the π-donating ability of the substituent on the carbene disfavors the isomerization (OMe>amide>phthalimide). | (23) |
 | (24) |
The reaction path found here for ethylene (and most likely representative of that of vinyl ether) shows the key role of the metal hydride in permiting the isomerization to occur. In the absence of the metal fragment a 1,2-shift of H in free ethylene is a very high energy process (>60 kcal mol−1 for propene10 and 79.1 kcal mol−10 for ethylene in these calculations). The presence of a transition metal complex has been shown to facilitate the 1,2 shift in the case of alkyne to vinylidene.14 Our calculations have shown that the multistep reaction initiated by alkyne insertion into the Ru–H bond is considerably lower in energy than the 1,2 shift. In the case of olefin, the 1,2 shift, even if also facilitated by the metal, should remain energetically inaccessible. The presence of an additional H on the metal is thus indispensable for the isomerization process.
The pathway presented here also explains the isotope labeling observation with C2D4. The isotope scrambling is explained by the olefin adduct entering the hydride migration path, even if accumulation of the carbene complex is not thermodynamically possible.
Since the final complex is unsaturated, a study of the effect of an electron-donating group remote from the olefinic function has been studied. It was hoped that this would stabilize the final carbene by coordination to the unsaturated site. With the exception of the amide function where the O is positioned in close proximity to the metal and thus has been shown to coordinate, it has been observed that a more remote functionality does not interact with the metal. This illustrates the relatively poor Lewis acidity of Ru with a hydride trans to the empty site.
The chemistry of transition metal carbene complexes, LnMC(X)R (i.e., terminal carbenes) has been categorized into two types, named after their discoverers as Fischer carbenes and Schrock carbenes. As outlined in a recent theoretical study,15 these are conventionally distinguished by a low metal oxidation state and X a π donor (OR′, NR2′, etc.) for Fischer carbenes, and a high metal oxidation state and X a pure sigma ligand (H, alkyl, silyl, etc.) for Schrock carbenes. In our case, is a Ru(II) complex of a carbene with a π-donor group RuHCl(C(OR)R′)L2 and no CO ligands a Fischer- or a Schrock-type carbene? These mentioned ‘‘distinguishing features’’ fail to recognize two other differences. (1) Fischer carbene complexes generally have the strong π acid CO for some or all of the co-ligands L (which will tend to minimize M→CXR back-bonding and thus put higher demands on X→C π bonding) while Schrock carbenes have strong sigma donors (alkyls and phosphines) and sometimes π donors (alkoxides) for L. (2) Fischer carbene complexes invariably have an 18-electron count at M while Schrock carbene complexes generally have 16 or fewer valence electrons. In short, it is unreasonable that the overall differences are based primarily on the MC(X)R substructure; the identity of L and the value of n in LnMC(X)R are important, and perhaps controlling factors.
Above and beyond this analysis of differences, a distinction between Fischer and Schrock carbenes is that the former react with the carbene carbon behaving like an electrophile, while Schrock carbenes react with the carbene carbon behaving like a nucleophile. It is thus paradoxical that the low oxidation state metal promotes electrophilic carbene character, since an electron-rich metal should maximize back donation. Likewise, a high metal oxidation state should not cause a nucleophilic carbene carbon. Moreover, it is also irrational that a π-donor substituent on the carbene carbon should leave that carbene electrophilic.
In addition, exceptions exist to the above systematics. Cp2Ta(CH3)(CH2) does not show Wittig reactivity under mild conditions. W(CPh2)(CO)5 does not effect olefin metathesis until one CO is displaced, from which we can conclude that olefin metathesis requires prior W–olefin bonding and does not proceed from direct contact between the carbene carbon and an olefin. Indeed, it is not at all established that olefin metathesis benefits from nucleophilic vs. electrophilic carbene carbon character. These characteristics may in fact be quite irrelevant.
Reactivity is not necessarily a reflection of the character of an unperturbed molecule. In particular, the Wittig-like reactivity of certain Schrock carbenes [eqn. (25), which is a prime source for the claim of nucleophilic carbon character in Schrock carbenes] probably originates from an adduct K in which the acetone carbon is made more electrophilic by Ta and such substrate binding also makes C′ more nucleophilic.
| R3TaCHR+Me2CO→‘‘R3TaO
’’+RHCCMe2 | (25) |
The unsaturation at M is probably a key factor in the reactivity dichotomy between Fischer and Schrock carbenes. In support of this assertion, a theoretical study15 has revealed how addition of one fluoride to the Schrock carbene model F4WCH2 to give F5WCH2−, alters the W–CH2 bond to something that resembles the same bond in (OC)5WCH2. Indeed, Roper and colleagues long ago noted that oxidation state is not a safe criterion for predicting electrophilic or nucleophilic reactivity.16 Two related CpRe(CO)2(CRR′) species are susceptible to both electrophilic and nucleophilic addition to the carbene carbon; when R is OEt, electrophilic addition (i.e., H+) no longer occurs.17 Thus, metal oxidation state is not a controlling factor, and unsaturation at the metal is a key (but not sufficient) factor. In this context, the ruthenium chemistry reported here illustrates a class of compounds that fit neither traditional carbene complex category. RuHCl[C(OR)R′]L2 carries a π-donor substituent on the carbene carbon, the metal is in an intermediate oxidation state [but Ru(II) is a fairly powerful π base], but the metal is unsaturated.

Several theoretical studies using GVB, NBO and CDA analyses have been carried out on representative selections of carbene complexes to classify the carbene complexes as a member of either Fischer or Schrock series.15 With these tools, Fischer carbenes were shown to be donor-acceptor complexes involving metal and carbene fragments in a singlet state, while the Schrock carbenes should be discussed in terms of interactions between metal and carbene fragments in the triplet states. If in fact the energy gap between singlet and triplet of each fragment is a controlling criterion, there is no reason to partition into two classes, as a continuum situation is highly probable. This is probably the right time to recognize that there will be a continuum of carbene ‘‘characters’’, and to cease trying to place new ones in either the Fischer or the Schrock category.17 The systems presented here offer an ideal study ground for a more in-depth understanding of what controls the changes from Fischer to Schrock carbenes. The presence of unsaturation and the nature of the ligand trans to the empty site are probably key factors that have not been noticed earlier.
A CDA analysis15 was carried out on several representative 16-electron carbene complexes resulting from union of the fragments RuXY(PH3)2 and CH(CH3) or C(OCH3)(CH3). As shown by this analysis, a Fischer complex is characterized by a large carbene→metal donation (large positive d), a large metal→carbene back donation (large positive b), a large repulsive polarization (large negative r) and a small residual ε. Schrock complexes are characterized mostly by large residual and small d, b and r. These values have been calculated (Table 5) for RuHCl(PH3)2(CH(CH3)), RuHCl(PH3)2(C(OCH3)(CH3)) in the three minima (7, 8 and 9) and for RuCl2(PH3)2(CH(CH3)) as a reference system. The parameter values show that the OCH3 substituted carbenes are ideal Fischer carbenes, especially in the two most stable conformations (7 and 8); conformation 9 has a slightly higher residual but is still clearly a Fischer carbene. RuCl2(PH3)2[CH(CH3)] is a typical Schrock complex with a large residual (0.325) and very small d, b and r values. Changing Cl into H on the metal gives a species that has reasonable d, b and r values but a non-negligible residual. It is thus remarkable that the same metal fragment with the same formal oxidation state can result in carbene complexes with a continuum of behavior between Fischer and Schrock types. This is probably associated with a metal fragment having a singlet-triplet gap highly sensitive to the nature of the substituent. This is apparently a very remarkable feature of the 14-electron RuXYL2 fragments.
| d | b | r | ε | |
|---|---|---|---|---|
| (H3P)2RuHCl[CH(CH3)] | 0.335 | 0.273 | −0.287 | 0.117 |
| (H3P)2RuHCl[C(OMe)(CH3)], 9 | 0.470 | 0.264 | −0.323 | 0.058 |
| (H3P)2RuHCl[C(OMe)(CH3)], 7 or 8 | 0.487 | 0.314 | −0.425 | 0.000 |
| (H3P)2RuCl2[CH(CH3)] | 0.003 | 0.058 | 0.008 | 0.325 |
The isomerization reaction type observed here seems quite
general for vinyl ethers; it demonstrates that the hydrido
carbene is more stable thermodynamically than either the
hydrido olefin or the alkyl isomer [eqn. (26)] and it shows that there is no tendency for the hetero substituent
on the carbene to diminish the unsaturation at Ru by direct O→Ru binding. Isotope labeling using CH2CD(OEt) and DFT calculations both prove that the regiochemistry of addition of Ru–H across the H2CCH(OR) bond is not selective, but that only
the direction shown in eqn. (26) leads to the thermodynamically preferred carbene. The alternative intermediate
[eqn. (27)] leads to a carbene devoid of heteroatom stabilization, which is apparently thermodynamically less stable.
 | (26) |
 | (27) |
Why has this facile isomerization route to carbene ligands not already been discovered? First, the organometallic chemistry of vinyl ethers has not been extensively investigated in modern times,18,19 and what has been done centers on electrophilic attack on the ether oxygen (i.e., RO− abstraction, to give vinyl complexes) of less electron-rich, and saturated molecules. More important, previous reports involved nonhydride compounds, which are thus unable to effect the mechanism established here. Finally, any previous study under hydrogen gas, even if it involved carbene intermediates, would have given an alkane product since the unsaturated carbenes of the sort produced here are readily hydrogenated to alkanes [eqn. (28); 1 atm H2 , C6D6, 60 min at 25°C]. To answer the question that begins this paragraph, a referee has commented that ‘‘It has, in a way . . . ’’ In a previous report,20 the thermodynamic preference for ethylidene in the isomerization of ethylene on a highly reducing (Me3SiNC2H4)3NTa fragment, was attributes to an α-agostic interaction.
 | (28) |
The implication of the above is that any
unsaturated monohydride has the potential to effect this carbene synthesis from
vinyl ethers. To be able to do this, the chosen metal must be
sufficiently electron-rich to tolerate the higher formal oxidation state of the carbene product. In practice, however, we have found that OsHCl(PPh3)3 can be stirred in C6H6 with equimolar H2CCH(OR) (R=Et or But) at 25°C for up to 90 h without change. With RuHCl(PPh3)3 there is no reaction with ethyl vinyl ether over 3 days at 60°C in C6D6. We continue to
search for other unsaturated monohydrides capable of this isomerization, with special attention to whether a 14-electron configuration and/or a four-coordinate structure is a necessity.
There is a general belief that Fischer carbenes are ‘‘more stable’’ than Schrock carbenes, and this is attributed to π donation by
the heteroatom to the carbene carbon. Quantitative comparison of ‘‘stability’’ is however not established in such commentaries
since there are no isomeric forms for experimental comparison. Instead, the discussion usually hinges on Fischer carbenes having an electrophlic carbon while Schrock carbenes have nucleophilic carbon; these have no direct relation to ‘‘stability
’’. What the calculations reveal here on the systems studied experimentally is that, for isomeric alternative carbenes on Ru, the heteroatom on the carbene carbon is indeed more stable. Via the thermodynamic cycles in Scheme 2, this can
be traced primarily to the properties of the free carbenes
[ΔE(α→β) vs. ΔE(β→α) to make the two different carbenes]. The heteroatom makes the
carbene more stable (more energetically accessible). Since
this is close to the calculated difference in overall ΔE(1) and
ΔE(2), it follows that the two BDEs do not differ greatly. That
is, differences in the bonding of the two isomeric carbenes
to Ru are less important than the differences in the energies
of the free
carbenes themselves. Thus, no comparative conclusions
can be drawn, from the observed isomerization, about the
RuC bond. The formation of the heteroatom carbene is possible
due to properties of the metal-free hydrocarbon isomers.
 | ||
| Scheme 2 | ||
Experimental
Computational details
Ab initio calculations were carried out with the Gaussian 9421 set of programs within the framework of DFT at the B3PW91 level.22 The Hay–Wadt effective core potential (quasi-relativistic for the metal centers) was used to replace the 28 innermost electrons of Ru23 and the 10 core electrons of P and Cl.24 The associated double ζ basis sets were used. They were augmented by d polarization functions for P and Cl.25 H, C, O and N were represented by a 6-31G(d,p) basis set.26 In the case of the olefin substituted by an amide group (10), a STO-3G basis set was used for the CH2 group of the five-membered ring as well as the H of PH3. Comparison between the results with the smaller basis set and the larger one (as defined above) for 11 proved the more restricted basis set to give good geometrical results. Full geometry optimization was performed without symmetry restriction. For the reaction path of isomerization of the C2H4 adduct into the C(H)CH3 complex, the nature of the optimized structure as a minimum or a transition state was checked by numerical frequency calculations. The transition-state structures were given small geometrical perturbations along the reaction coordinates and then further geometrical optimization was carried out to ensure they connect the reactant and product of interest.Syntheses and reactions
All manipulations were performed using standard Schlenk techniques or in an argon-filled glovebox unless otherwise noted. Solvents were distilled from Na/benzophenone or CaH2 , degassed prior to use, and stored in air-tight vessels. Commercially available vinyl ethers, amines and amides were used as received after drying and degassing. 1H NMR chemical shifts are reported in ppm relative to protio impurities in the deutero solvents. 31P and 19F NMR spectra are referenced to external standards of 85% H3PO4 and CFCl3, respectively (both at 0 ppm). NMR spectra were recorded with either Varian GEMINI 2000 (300 MHz 1H; 121 MHz 31P; 75 MHz 13C; 282 MHz 19F) or Varian UNITY INOVA (400 MHz 1H; 162 MHz 31P; 101 MHz 13C; 376 MHz 19F) instruments. IR spectra were recorded on a Nicolet 510P FT-IR spectrometer equipped with OMNIC v.4.1b software.![[double bond, length half m-dash]](https://www.rsc.org/images/entities/b_char_e006.gif) CH2)..
Under Ar, 10 mg (0.022 mmol)
RuHCl(PPr3i)2 was added to an NMR tube equipped with a
Teflon stopcock. The tube was filled with C6D6 to a predetermined mark on the tube so that the remaining head space
volume was 4.0 mL (approximately 0.5 mL C6D6). The NMR
tube was cooled to O°C to freeze the benzene-d6 and was then evacuated. It was then filled with ethylene (0.022 mmol) to 95 mm Hg and the stopcock closed. The sample was then warmed to room temperature, shaken, and its 1H and 31P{1H}
NMR spectra taken over 30 min intervals. The adduct that formed showed little or no decomposition over a period of several hours. 1H NMR (400 MHz, C6D6, 20°C): δ
−22.0 (t, 2JP-H=18.0 Hz, RuH), 1.15 [dvt, JP-H=3JH-H=6.0 Hz, 18H, P(CHMe2)3],
1.20 [dvt, JP-H=3JH-H=6.0 Hz, 18H, P(CHMe2)3], 2.25 [m, 6H, P(CHMe2)3],
2.79 [t, 4JP-H=3.2 Hz, 4H, Ru(CH2CH2)]. 31P{1H} NMR
(162 MHz, C6D6, 20°C): δ 44.1 (s).CD2. Reversible insertion.. Under Ar, 10 mg (0.022 mmol) RuHCl(PPr3i)2 was
added to an NMR tube equipped with a Teflon stopcock in
0.5 mL of benzene. The tube was frozen in ice, evacuated, and
CD2CD2 (600 torr) was added. 2H NMR (20°C) spectra taken
after 1 h at room temperature show deuterium incorporation into the PPr3i methyl groups (δ 1.2), and into the hydride position
(δ
−22.0) in addition to being present in the olefinic adduct (δ 2.8). This is indicative of reversible insertion of the ethylene unit into the Ru–H bond.C(Me)OEt.. Under Ar, 25 mg (0.055 mmol)
of RuHCl(PPr3i)2 was placed in an NMR tube in C6D6. Via
syringe, 5.2 μL (0.055 mmol) of ethyl vinyl ether was added and the NMR tube sealed. 1H, 13C{1H}, and 31P{1H} NMR spectra taken after approximately 30 min revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OEt). The complex can be isolated as an orange-brown viscous oil from benzene if a larger quantity is required. 1H NMR (300 MHz, C6D6
, 20°C): δ
−21.65 (t, 2JP-H=22.8 Hz, 1H, RuH), 1.13 [dvt, JP-H=3JH-H=6.3 Hz, 18H0, P(CHMe2)3],
1.25 [dvt, JP-H=3JH-H=6.3 Hz, 18H, P(CHMe2)3], 1.17
[t, 3JH-H=7.2 Hz, 3H, RuC(Me)OCH2CH3], 2.33–2.44 [m, 6H, P(CHMe2)3],
2.69 [s, 3H, RuC(Me)OEt], 4.37 [q, 3JH-H=7.5 Hz, 2H,
RuC(Me)OCH2CH3]. 31P{1H} NMR (121 MHz, C6D6, 20°C): δ 58.2 (s). 13C{1H} NMR (75.5 MHz, C6D6, 20°C): δ 14.6 [s, RuC(Me)OCH2CH3], 19.7 [s, P(CHMe2)3], 20.4 [s, P(CHMe2)3], 26.2 [vt, JP-C=9.1
Hz, P(CHMe2)3], 40.8 [s, RuC(Me)OEt], 68.5 [s, RuC(Me)OCH2CH3], 289.8
[t, 2JP-C=9.7 Hz, RuC(Me)OEt].
CH2)..
Under Ar, 10 mg (0.022 mmol)
RuHCl(PPr3i)2 was added to an NMR tube equipped with a
Teflon stopcock. The tube was filled with C6D6 to a predetermined mark on the tube so that the remaining head space
volume was 4.0 mL (approximately 0.5 mL C6D6). The NMR
tube was cooled to O°C to freeze the benzene-d6 and was then evacuated. It was then filled with ethylene (0.022 mmol) to 95 mm Hg and the stopcock closed. The sample was then warmed to room temperature, shaken, and its 1H and 31P{1H}
NMR spectra taken over 30 min intervals. The adduct that formed showed little or no decomposition over a period of several hours. 1H NMR (400 MHz, C6D6, 20°C): δ
−22.0 (t, 2JP-H=18.0 Hz, RuH), 1.15 [dvt, JP-H=3JH-H=6.0 Hz, 18H, P(CHMe2)3],
1.20 [dvt, JP-H=3JH-H=6.0 Hz, 18H, P(CHMe2)3], 2.25 [m, 6H, P(CHMe2)3],
2.79 [t, 4JP-H=3.2 Hz, 4H, Ru(CH2CH2)]. 31P{1H} NMR
(162 MHz, C6D6, 20°C): δ 44.1 (s).CD2. Reversible insertion.. Under Ar, 10 mg (0.022 mmol) RuHCl(PPr3i)2 was
added to an NMR tube equipped with a Teflon stopcock in
0.5 mL of benzene. The tube was frozen in ice, evacuated, and
CD2CD2 (600 torr) was added. 2H NMR (20°C) spectra taken
after 1 h at room temperature show deuterium incorporation into the PPr3i methyl groups (δ 1.2), and into the hydride position
(δ
−22.0) in addition to being present in the olefinic adduct (δ 2.8). This is indicative of reversible insertion of the ethylene unit into the Ru–H bond.C(Me)OEt.. Under Ar, 25 mg (0.055 mmol)
of RuHCl(PPr3i)2 was placed in an NMR tube in C6D6. Via
syringe, 5.2 μL (0.055 mmol) of ethyl vinyl ether was added and the NMR tube sealed. 1H, 13C{1H}, and 31P{1H} NMR spectra taken after approximately 30 min revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OEt). The complex can be isolated as an orange-brown viscous oil from benzene if a larger quantity is required. 1H NMR (300 MHz, C6D6
, 20°C): δ
−21.65 (t, 2JP-H=22.8 Hz, 1H, RuH), 1.13 [dvt, JP-H=3JH-H=6.3 Hz, 18H0, P(CHMe2)3],
1.25 [dvt, JP-H=3JH-H=6.3 Hz, 18H, P(CHMe2)3], 1.17
[t, 3JH-H=7.2 Hz, 3H, RuC(Me)OCH2CH3], 2.33–2.44 [m, 6H, P(CHMe2)3],
2.69 [s, 3H, RuC(Me)OEt], 4.37 [q, 3JH-H=7.5 Hz, 2H,
RuC(Me)OCH2CH3]. 31P{1H} NMR (121 MHz, C6D6, 20°C): δ 58.2 (s). 13C{1H} NMR (75.5 MHz, C6D6, 20°C): δ 14.6 [s, RuC(Me)OCH2CH3], 19.7 [s, P(CHMe2)3], 20.4 [s, P(CHMe2)3], 26.2 [vt, JP-C=9.1
Hz, P(CHMe2)3], 40.8 [s, RuC(Me)OEt], 68.5 [s, RuC(Me)OCH2CH3], 289.8
[t, 2JP-C=9.7 Hz, RuC(Me)OEt].Upon cooling a sample of RuHCl(PPr3i)2(C(Me)OEt) in toluene-d8
to −90°C, slowed rotation of the ruthenium carbene
bond allowed detection of the two isomers, which differ in E/Z stereochemistry about the (X)(Y)RuCRR′ bond. 1H NMR (400 MHz, toluene-d8, −80°C): New signals were seen at δ 4.95 and
4.29 [1:10 ratio, coalesced to the time-averaged signal at δ 4.38, RuC(Me)OCH2CH3], δ 2.06 and 2.87 (1:10 ratio, coalesce at δ 2.68, RuC(Me)OCH2CH3],
and δ
−25.38 and −21.29 (1:10 ratio, coalesce at δ
−21.68, RuH).
31P{1H} NMR (162 MHz, toluene-d8, −80°C): δ 51.8 and 56.4 (1:10 ratio, coalesce
at
δ 56.0).
CHOEt) follows. The olefinic
protons of the adduct were not resolved due to coincidental
overlap with the PPr3i
ligands. 1H NMR (300 MHz, toluene-d8):
δ
−15.52 (t, 2JP-H=17.7 Hz, 1H, RuH), 3.69 [m, 1H, Ru(CH2CHOCH2CH3)],
4.35 [m, 1H, Ru(CH2CHOCH2CH3)], 2.02 [m, 3H, P(CHMe2)3],
2.32 [m, 3H, P(CHMe2)3]. 31P{1H} NMR (121 MHz, toluene-d8):
δ 40.8, 46.0 (AB pattern, 2JPA–PB=300 Hz).C(OEt)Me).. Under argon,
10.0 mg (0.022 mmol) RuHClL2 was dissolved in 0.5 mL
C6D6 in an NMR tube equipped with a Teflon stopcock and
2.4 μL (0.022 mmol) ethyl vinyl ether added. After formation of the
carbene, as verified by 31P NMR, the solution was frozen
in ice, the headspace gases removed, and the tube filled to 1
atm with H2. After 30 min. at room temperature, over 50% conversion of RuHClL2(C(OEt)Me) to RuH3ClL228 and diethyl
ether was achieved.![[upper bond 1 start]](https://www.rsc.org/images/entities/b_char_e010.gif) OCH2CH2C
OCH2CH2C![[upper bond 1 end]](https://www.rsc.org/images/entities/b_char_e011.gif) H2).. Under
Ar, 25 mg (0.055
mmol) RuHCl(PPr3i)2 was added to an NMR tube in C6D6.
Via syringe, 4.2 μL (0.055 mmol) of 2,3-dihydrofuran was added
and the tube sealed. After 30 min NMR spectra were recorded. The benzene-d6 was then removed to a −196°C trap, the
product was re-dissolved in CD2Cl2
, and the 1H spectra measured again to resolve all peaks. In C6D6, the central methylene
unit of the carbene substituent was not resolved.
1H, 13C{1H}, and 31P{1H} NMR showed quantitative conversion to RuHCl(PPr3i)2(C
H2).. Under
Ar, 25 mg (0.055
mmol) RuHCl(PPr3i)2 was added to an NMR tube in C6D6.
Via syringe, 4.2 μL (0.055 mmol) of 2,3-dihydrofuran was added
and the tube sealed. After 30 min NMR spectra were recorded. The benzene-d6 was then removed to a −196°C trap, the
product was re-dissolved in CD2Cl2
, and the 1H spectra measured again to resolve all peaks. In C6D6, the central methylene
unit of the carbene substituent was not resolved.
1H, 13C{1H}, and 31P{1H} NMR showed quantitative conversion to RuHCl(PPr3i)2(C![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) OCH2CH2CH2). 1H NMR (300 MHz,
CD2Cl2, 20°C): δ
−18.20 (t, 2JP-H=21.9 Hz, 1H, RuH), 1.19 [dvt,
JP-H=3JH-H=6.3 Hz, 18H, P(CHMe2)3], 1.21 [dvt, JP-H=3JH-H=6.3
Hz, 18H, P(CHMe2)3], 1.76 (apparent quintet,
3JH-H=7.3 Hz, 2H, RuCOCH2CH2CH2), 2.27–2.34 [m, 6H, P(CHMe2)3],
3.07 (t, 3JH-H=7.7 Hz, 2H, RuCOCH2CH2CH2), 4.19 (t, 3JH-H=6.9
Hz, 2H, RuCOCH2CH2CH2). 31P{1H} NMR (121 MHz, C6D6, 20°C):
δ 60.1 (s). 13C{1H} NMR (75.5 MHz, C6D6, 20°C): δ 20.5
[s, P(CHMe2)3], 20.8 [s, P(CHMe2)3], 24.0 (s, RuCOCH2CH2CH2),
25.5 [vt, JP-C=9.4 Hz, P(CHMe2)3], 51.4 (s, RuCOCH2CH2CH2), 76.0 (s, RuCOCH2CH2CH2), 286.6 (t, 2JP-C=9.4 Hz, RuCOCH2CH2CH2).C(Me)OC6H11).. Under Ar, 10 mg (0.022
mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 3.1 μL (0.022 mmol) of cyclohexyl vinyl ether was added and the tube sealed. 1H and 31P{1H} NMR spectra taken after 1 h showed complete conversion to RuHCl(PPr3i)2(C(Me)OC6H11).
1H NMR (400 MHz, C6D6, 20°C): δ
−19.39 (t, 2JP-H=22.8 Hz, 1H, RuH), 1.21 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3],
1.33 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3],
1.03–1.75 [m, 10H, RuC(Me)OC6H11], 2.40–2.49 [m, 6H, P(CHMe2)3], 2.72 [s, 3H, RuC(Me)OC6H11], 4.38–4.46 [m, 1H, RuC(Me)OC6H11]. 31P{1H} NMR (162 MHz, C6D6, 20°C): δ 57.4 (s).C(Me)OSiMe3).. Under Ar, 20 mg (0.044
mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 6.7 μL (0.044 mmol) of vinyloxytrimethylsilane was added and the tube sealed. 1H, 13C{1H}, and 31P{1H} NMR spectra taken after 2 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OSiMe3). 1H NMR (400 MHz, C6D6, 20°C): δ
−17.96 (t, 2JP-H=22.0 Hz, 1H, RuH), 0.03 [s, 9H, RuC(Me)OSiMe3], 1.24 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3],
1.27 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 2.30–2.40 [m,
6H, P(CHMe2)3], 2.57 [s, 3H, RuC(Me)OSiMe3]. 31P{1H}
NMR (162 MHz, C6D6, 20°C): δ 58.6 (s). 13C{1H} NMR (101 MHz, C6D6,
20°C): δ 1.2 [s, RuC(Me)OSiMe3], 20.2 [s, P(CHMe2)3], 20.4
[s, P(CHMe2)3], 25.3 [vt, JP-C=9.5 Hz, P(CHMe2)3], 43.1 [s, RuC(Me)OSiMe3],
283.8 [t, 2JP-C=8.4 Hz, RuC(Me)OSiMe3].C(Me)OCH2CH2OBun).. Under Ar, 20 mg
(0.044 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 7.3 μL (0.044 mmol) of ethylene glycol butyl vinyl ether was added and the tube sealed. 1H, 13C{1H} and 31P{1H}
NMR spectra taken after 24 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2OBun). 1H NMR (300 MHz,
C6D6, 20°C): δ
−21.29 (t, 2JP-H=21.6 Hz, 1H, RuH), 0.86 (t,
3JH-H=7.5 Hz, 3H, OCH2CH2CH2CH3), 1.07 (apparent
sextet, 3JH-H=7.8 Hz, 2H, OCH2CH2CH2CH3), 1.17 [dvt,
JP-H=3JH-H=6.6 Hz, 18H, P(CHMe2)3], 1.30 [dvt, JP-H=3JH-H=6.6
Hz, 18H, P(CHMe2)3], 1.49 (apparent quintet, 3JH-H=7.8
Hz, 2H, OCH2CH2CH2CH3), 2.40–2.50 [m, 6H, P(CHMe2)3], 2.76 [s, 3H, RuC(Me)OR], 3.27 (t, 3JH-H=6.9 Hz, 2H, OCH2CH2CH2CH3),
3.56 (t, 3JH-H=4.0 Hz,
2H, OCH2CH2OBun),
4.56 (t, 3JH-H=4.0 Hz, 2H, OCH2CH2OBun). 31P{1H}
NMR (121 MHz, C6D6, 20°C): δ 57.8 (s). 13C{1H}
NMR (75.5 MHz, C6D6, 20°C): δ 14.3 (s, OCH2CH2CH2CH3), 19.9
(s, OCH2CH2CH2CH3), 20.0 [s, P(CHMe2)3], 20.7 [s, P(CHMe2)3], 26.5
[vt, JP-C=9.5 Hz, P(CHMe2)3], 32.4 (s, OCH2CH2CH2CH3), 40.8
[s, RuC(Me)OR], 69.5 (s, OCH2CH2CH2CH3), 71.4 (s, OCH2CH2OBun),
72.7 (s, OCH2CH2OBun), 288.7 (t, 2JP-C=9.5 Hz, RuC).C(Me)OCH2CH2OH).. Under Ar, 25 mg
(0.055 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 4.9 μL (0.055 mmol) of ethylene glycol vinyl ether was added and the tube sealed. 1H, 13C{1H}, and 31P{1H} NMR spectra taken after 6 h revealed conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2OH).
1H NMR (400 MHz, C6D6, 20°C): δ
−21.11 (t, 2JP-H=21.6 Hz, 1H, RuH), 1.13 [dvt, JP-H=3JH-H=6.6 Hz, 18H, P(CHMe2)3], 1.26 [dvt, JP-H=3JH-H=6.6 Hz, 18H, P(CHMe2)3], 2.36–2.46 [m, 6H, P(CHMe2)3], 2.72 [s, 3H, RuC(Me)OR], 3.53 (s, 1H, OH) when 10.0 mg RuHClL2 and equimolar olefin are used in the same solvent volume, OH has δ 3.36); 3.68 (t,
3JH-H=4.8 Hz, 2H, OCH2CH2OH), 4.36 (t, 3JH-H=4.8 Hz, 2H, OCH2CH2OH). 31P{1H} NMR (162 MHz, C6D6, 20°C): δ 56.4 (s). 13C{1H} NMR (101 MHz, C6D6, 20°C): δ 19.7 [s, P(CHMe2)3], 20.4 [s, P(CHMe2)3], 26.3 [vt, JP-C=10.0 Hz, P(CHMe2)3]), 40.8 [s,
RuC(Me)OR], 61.1 (s, OCH2CH2OH), 74.3 (s, OCH2CH2OH), 288.8 (t, 2JP-C=8.4 Hz, RuC).C(Me)OCH2CH2OCH2CH2OH).. Under
Ar, 10 mg (0.022 mmol) of RuHCl(PPr3i)2 was placed in an
NMR tube in C6D6. Via syringe, 3.0 μL (0.022 mmol) of diethylene glycol vinyl ether was added and the tube sealed. 1H and 31P{1H} NMR spectra taken after 1 h revealed conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2OCH2CH2OH). 1H NMR (400
MHz, C6D6, 20°C): δ
−21.40 (t, 2JP-H=21.2 Hz, 1H, RuH), 1.15 [dvt, JP-H=3JH-H=6.8 Hz, 18H, P(CHMe2)3], 1.27 [dvt, JP-H=3JH-H=6.8
Hz, 18H, P(CHMe2)3], 2.37–2.47 [m, 6H, P(CHMe2)3],
2.71 [s, 3H, RuC(Me)OR], δ 3.25 (t, 3JH-H=4.4 Hz, 2H,
OCH2CH2OH), 3.51 (m, 4H, CH2OCH2), 4.51 [t, 3JH-H=4.4 Hz, 2H, RuC(Me)OCH2], the OH proton is obscured by signals for the protons vicinal to the ether oxygen(s). 31P{1H} NMR (162
MHz, C6D6, 20°C): δ 56.3 (s).C(Me)OCH2CH2O(Me)C)RuHCl(PPr3i)2..
Under Ar, 150 mg (0.328 mmol) of RuHCl(PPr3i)2 was charged
in a Schlenk flask and dissolved in 10 mL of benzene. Over a
period of 1 h, 20.5 μL (0.164 mmol) of ethylene glycol divinyl ether in 5 mL of benzene was added through a pressure equalized
dropping funnel and the reaction stirred overnight. The benzene was removed to a N2 trap and the residue was dissolved in pentane. Cooling at −60°C overnight afforded a brown precipitate that was washed with a small amount of cold pentane and dried in vacuo. Isolated yield: 65 mg (90% by 31P{1H} NMR integration before work-up). 1H, 13C{1H}, and 31P{1H} NMR showed clean conversion to [RuHCl(PPr3i)2(C(Me)OCH2–)]2.
1H NMR (300 MHz, C6D6, 20°C): δ
−21.21 (t,
2JP-H=21.8 Hz, 1H, RuH), 1.17 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.30 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], δ 2.33–2.44 [m, 6H, P(CHMe2)3], 2.73 [s, 3H, RuC(Me)OCH2–], 4.85 [s, 2H, RuC(Me)OCH2–]. 31P{1H} NMR (121 MHz, C6D6, 20°C):
δ 57.9 (s). 13C{1H} NMR (101 MHz, C6D6, 20°C): δ 19.7 [s, P(CHMe2)3], 20.5 [s,
P(CHMe2)3], 26.3 [vt, JP-C=9.7 Hz, P(CHMe2)3], 41.3 [s, RuC(Me)OCH2–],
71.3 [s, RuC(Me)OCH2–], 288.8 (t, 2JP-C=9.1 Hz, RuC(Me)OCH2–].CH(OCH2-CH2F).. Under
Ar, 1.85 g (11.5 mmol) of diethylamino sulfur
trifluoride (DAST)29 was dissolved in 10 mL CH2Cl2. The
solution was cooled to −78°C and 1.03 mL (11.5 mmol) of ethylene glycol vinyl ether was added via syringe. The solution was allowed to warm to room temperature and stirred for 1 h, then cooled to 0°C and the solvent removed to an N2 trap. The product was then fractionally distilled, collecting the 79–81°C
fraction (760 torr). Yield : approx. 500 mg (49%). 1H NMR (300 MHz, C6D6, 20°C):δ 3.28 (dt, 3JF-H=28.5 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F), 3.89 [dd, 3JH-H=6.9 Hz, 2JH-H=1.5 Hz, 1H, CH2CH(OR)], 4.01 [dd, 3JH-H=14.1 Hz, 2JH-H=1.5 Hz, 1H, CH2CH(OR)], 4.06 (dt, 3JF-H=47.7 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F), 6.30
[dd, 3JH-H=14.1 Hz, 3JH-H=6.9 Hz, 1H, CH2CH(OR)]. 13C{1H} NMR
(75.5 MHz, C6D6, 20°C): δ 67.0 (d, 2JF-C=20.5 Hz, OCH2CH2F),
81.5 (d, 1JF-C=171 Hz, OCH2CH2F), 86.1 [s, CH2CH(OR)], 151.7 [s,
CH2CH(OR)]. 19F NMR (282 MHz, C6D6, 20°C): δ
−224.8 (tt, 2JH-F=47.7
Hz, 3JH-F=28.5 Hz).C(Me)OCH2CH2F).. Under Ar, 10 mg
(0.022 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 2.5 μL (0.022 mmol) of 2-fluoroethyl vinyl ether
was added and the NMR tube sealed. 1H, 19F, and 31P{1H}
NMR spectra taken after 3 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2F). 1H NMR (300 MHz, C6D6, 20°C): δ
−21.32 (t, 2JP-H=21.6 Hz, 1H, RuH), 1.12 [dvt, JP-H=3JH-H=6.3
Hz, 18H, P(CHMe2)3], 1.25 [dvt, JP-H=3JH-H=6.3 Hz, 18H, P(CHMe2)3], 2.32–2.44 [m, 6H, P(CHMe2)3], 2.72 [s, 3H, RuC(Me)OR], 4.26 (dt, 2JF-H=47.7 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F), 4.46 (dt, 3JF-H=29.7 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F). 31P{1H} NMR (121 MHz, C6D6, 20°C): δ 57.8 (s). 19F NMR (282 MHz, C6D6, 20°C): δ
−224.1 (tt, 2JH-F=47.7 Hz, 3JHF=29.7 Hz).C(Me)OCH2CH2NEt2).. Under Ar, 10 mg
(0.022 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 4.1 μL (0.022 mmol) of 2-(diethylamino)ethanol vinyl ether was added and the tube sealed. 1H and 31P{1H}
NMR spectra taken after 90 min revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2NEt2). 1H NMR (400 MHz, C6D6, 20°C): δ
−21.41 (brs, 1H, RuH), 0.96 [t, 3JH-H=7.2 Hz,
3H, N(CH2CH3)2], 1.16 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.29 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 2.40–2.50 (m,
6H, P(CHMe2)3], 2.47 [q, 3JH-H=7.2 Hz, 2H, N(CH2CH3)2], 2.74 [s, 3H, RuC(Me)OR], 2.77 (t, 3JH-H=6.8 Hz, 2H, OCH2CH2NEt2), 4.54 (t, 3JH-H=6.8 Hz, 2H, OCH2CH2NEt2). 31P{1H} NMR (162 MHz, C6D6, 20°C): δ 56.4 (s).C(Me)OCH2CHOCH2).. Under Ar, 10 mg
(0.022 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 2.2 μL (0.022 mmol) of glycidyl vinyl ether (racemic) was added and the NMR tube sealed. 1H and 31P{1H} NMR spectra taken after 1 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CHOCH2). 1H NMR (300 MHz,
C6D6, 20°C): δ
−21.41 (t, 2JP-H=22.2 Hz, 1H, RuH), 1.15 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.26 [dvt, JP-H=3JH-H=6.4
Hz, 18H, P(CHMe2)3], 2.22 (dd, JH-H=5.3, 2.3
Hz, 1H, OCH2CHOCH2), 2.30 (apparent t, JH-H=4.4
Hz, 1H, OCH2CHOCH2), 2.35–2.45 [m, 6H, P(CHMe2)3],
2.72 [s, 3H, RuC(Me)OR], 3.00 (m, 1H, OCH2CHOCH2), 4.34
(dd, JH-H=13.4, 6.3 Hz, 1H, OCH2CHOCH2), 4.58 (dd, JH-H=13.4,
2.1 Hz, 1H, OCH2CHOCH2). 31P{1H} NMR (121 MHz,
C6D6, 20°C): δ 56.33, 56.24 (AB pattern, 2JP-P=220 Hz).HCl(PPr3i)2(C(Me)N(Me)C(O)CH3).. Under
Ar, 75 mg
(0.164 mmol) of RuHCl(PPr3i)2 was charged in a Schlenk flask
and dissolved in 10 mL of benzene. Via syringe, 18.6 μL (0.180 mmol) of N-methyl-N-vinylacetamide was added and the solution stirred for 4 days. The benzene was removed to a N2 trap and the purple solid was washed with pentane. Drying in
vacuo yielded 40 mg (45%) of RuHCl(PPr3i)2(C(Me)N(Me)C(O
OCH2CH2CH2). 1H NMR (300 MHz,
CD2Cl2, 20°C): δ
−18.20 (t, 2JP-H=21.9 Hz, 1H, RuH), 1.19 [dvt,
JP-H=3JH-H=6.3 Hz, 18H, P(CHMe2)3], 1.21 [dvt, JP-H=3JH-H=6.3
Hz, 18H, P(CHMe2)3], 1.76 (apparent quintet,
3JH-H=7.3 Hz, 2H, RuCOCH2CH2CH2), 2.27–2.34 [m, 6H, P(CHMe2)3],
3.07 (t, 3JH-H=7.7 Hz, 2H, RuCOCH2CH2CH2), 4.19 (t, 3JH-H=6.9
Hz, 2H, RuCOCH2CH2CH2). 31P{1H} NMR (121 MHz, C6D6, 20°C):
δ 60.1 (s). 13C{1H} NMR (75.5 MHz, C6D6, 20°C): δ 20.5
[s, P(CHMe2)3], 20.8 [s, P(CHMe2)3], 24.0 (s, RuCOCH2CH2CH2),
25.5 [vt, JP-C=9.4 Hz, P(CHMe2)3], 51.4 (s, RuCOCH2CH2CH2), 76.0 (s, RuCOCH2CH2CH2), 286.6 (t, 2JP-C=9.4 Hz, RuCOCH2CH2CH2).C(Me)OC6H11).. Under Ar, 10 mg (0.022
mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 3.1 μL (0.022 mmol) of cyclohexyl vinyl ether was added and the tube sealed. 1H and 31P{1H} NMR spectra taken after 1 h showed complete conversion to RuHCl(PPr3i)2(C(Me)OC6H11).
1H NMR (400 MHz, C6D6, 20°C): δ
−19.39 (t, 2JP-H=22.8 Hz, 1H, RuH), 1.21 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3],
1.33 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3],
1.03–1.75 [m, 10H, RuC(Me)OC6H11], 2.40–2.49 [m, 6H, P(CHMe2)3], 2.72 [s, 3H, RuC(Me)OC6H11], 4.38–4.46 [m, 1H, RuC(Me)OC6H11]. 31P{1H} NMR (162 MHz, C6D6, 20°C): δ 57.4 (s).C(Me)OSiMe3).. Under Ar, 20 mg (0.044
mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 6.7 μL (0.044 mmol) of vinyloxytrimethylsilane was added and the tube sealed. 1H, 13C{1H}, and 31P{1H} NMR spectra taken after 2 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OSiMe3). 1H NMR (400 MHz, C6D6, 20°C): δ
−17.96 (t, 2JP-H=22.0 Hz, 1H, RuH), 0.03 [s, 9H, RuC(Me)OSiMe3], 1.24 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3],
1.27 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 2.30–2.40 [m,
6H, P(CHMe2)3], 2.57 [s, 3H, RuC(Me)OSiMe3]. 31P{1H}
NMR (162 MHz, C6D6, 20°C): δ 58.6 (s). 13C{1H} NMR (101 MHz, C6D6,
20°C): δ 1.2 [s, RuC(Me)OSiMe3], 20.2 [s, P(CHMe2)3], 20.4
[s, P(CHMe2)3], 25.3 [vt, JP-C=9.5 Hz, P(CHMe2)3], 43.1 [s, RuC(Me)OSiMe3],
283.8 [t, 2JP-C=8.4 Hz, RuC(Me)OSiMe3].C(Me)OCH2CH2OBun).. Under Ar, 20 mg
(0.044 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 7.3 μL (0.044 mmol) of ethylene glycol butyl vinyl ether was added and the tube sealed. 1H, 13C{1H} and 31P{1H}
NMR spectra taken after 24 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2OBun). 1H NMR (300 MHz,
C6D6, 20°C): δ
−21.29 (t, 2JP-H=21.6 Hz, 1H, RuH), 0.86 (t,
3JH-H=7.5 Hz, 3H, OCH2CH2CH2CH3), 1.07 (apparent
sextet, 3JH-H=7.8 Hz, 2H, OCH2CH2CH2CH3), 1.17 [dvt,
JP-H=3JH-H=6.6 Hz, 18H, P(CHMe2)3], 1.30 [dvt, JP-H=3JH-H=6.6
Hz, 18H, P(CHMe2)3], 1.49 (apparent quintet, 3JH-H=7.8
Hz, 2H, OCH2CH2CH2CH3), 2.40–2.50 [m, 6H, P(CHMe2)3], 2.76 [s, 3H, RuC(Me)OR], 3.27 (t, 3JH-H=6.9 Hz, 2H, OCH2CH2CH2CH3),
3.56 (t, 3JH-H=4.0 Hz,
2H, OCH2CH2OBun),
4.56 (t, 3JH-H=4.0 Hz, 2H, OCH2CH2OBun). 31P{1H}
NMR (121 MHz, C6D6, 20°C): δ 57.8 (s). 13C{1H}
NMR (75.5 MHz, C6D6, 20°C): δ 14.3 (s, OCH2CH2CH2CH3), 19.9
(s, OCH2CH2CH2CH3), 20.0 [s, P(CHMe2)3], 20.7 [s, P(CHMe2)3], 26.5
[vt, JP-C=9.5 Hz, P(CHMe2)3], 32.4 (s, OCH2CH2CH2CH3), 40.8
[s, RuC(Me)OR], 69.5 (s, OCH2CH2CH2CH3), 71.4 (s, OCH2CH2OBun),
72.7 (s, OCH2CH2OBun), 288.7 (t, 2JP-C=9.5 Hz, RuC).C(Me)OCH2CH2OH).. Under Ar, 25 mg
(0.055 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 4.9 μL (0.055 mmol) of ethylene glycol vinyl ether was added and the tube sealed. 1H, 13C{1H}, and 31P{1H} NMR spectra taken after 6 h revealed conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2OH).
1H NMR (400 MHz, C6D6, 20°C): δ
−21.11 (t, 2JP-H=21.6 Hz, 1H, RuH), 1.13 [dvt, JP-H=3JH-H=6.6 Hz, 18H, P(CHMe2)3], 1.26 [dvt, JP-H=3JH-H=6.6 Hz, 18H, P(CHMe2)3], 2.36–2.46 [m, 6H, P(CHMe2)3], 2.72 [s, 3H, RuC(Me)OR], 3.53 (s, 1H, OH) when 10.0 mg RuHClL2 and equimolar olefin are used in the same solvent volume, OH has δ 3.36); 3.68 (t,
3JH-H=4.8 Hz, 2H, OCH2CH2OH), 4.36 (t, 3JH-H=4.8 Hz, 2H, OCH2CH2OH). 31P{1H} NMR (162 MHz, C6D6, 20°C): δ 56.4 (s). 13C{1H} NMR (101 MHz, C6D6, 20°C): δ 19.7 [s, P(CHMe2)3], 20.4 [s, P(CHMe2)3], 26.3 [vt, JP-C=10.0 Hz, P(CHMe2)3]), 40.8 [s,
RuC(Me)OR], 61.1 (s, OCH2CH2OH), 74.3 (s, OCH2CH2OH), 288.8 (t, 2JP-C=8.4 Hz, RuC).C(Me)OCH2CH2OCH2CH2OH).. Under
Ar, 10 mg (0.022 mmol) of RuHCl(PPr3i)2 was placed in an
NMR tube in C6D6. Via syringe, 3.0 μL (0.022 mmol) of diethylene glycol vinyl ether was added and the tube sealed. 1H and 31P{1H} NMR spectra taken after 1 h revealed conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2OCH2CH2OH). 1H NMR (400
MHz, C6D6, 20°C): δ
−21.40 (t, 2JP-H=21.2 Hz, 1H, RuH), 1.15 [dvt, JP-H=3JH-H=6.8 Hz, 18H, P(CHMe2)3], 1.27 [dvt, JP-H=3JH-H=6.8
Hz, 18H, P(CHMe2)3], 2.37–2.47 [m, 6H, P(CHMe2)3],
2.71 [s, 3H, RuC(Me)OR], δ 3.25 (t, 3JH-H=4.4 Hz, 2H,
OCH2CH2OH), 3.51 (m, 4H, CH2OCH2), 4.51 [t, 3JH-H=4.4 Hz, 2H, RuC(Me)OCH2], the OH proton is obscured by signals for the protons vicinal to the ether oxygen(s). 31P{1H} NMR (162
MHz, C6D6, 20°C): δ 56.3 (s).C(Me)OCH2CH2O(Me)C)RuHCl(PPr3i)2..
Under Ar, 150 mg (0.328 mmol) of RuHCl(PPr3i)2 was charged
in a Schlenk flask and dissolved in 10 mL of benzene. Over a
period of 1 h, 20.5 μL (0.164 mmol) of ethylene glycol divinyl ether in 5 mL of benzene was added through a pressure equalized
dropping funnel and the reaction stirred overnight. The benzene was removed to a N2 trap and the residue was dissolved in pentane. Cooling at −60°C overnight afforded a brown precipitate that was washed with a small amount of cold pentane and dried in vacuo. Isolated yield: 65 mg (90% by 31P{1H} NMR integration before work-up). 1H, 13C{1H}, and 31P{1H} NMR showed clean conversion to [RuHCl(PPr3i)2(C(Me)OCH2–)]2.
1H NMR (300 MHz, C6D6, 20°C): δ
−21.21 (t,
2JP-H=21.8 Hz, 1H, RuH), 1.17 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.30 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], δ 2.33–2.44 [m, 6H, P(CHMe2)3], 2.73 [s, 3H, RuC(Me)OCH2–], 4.85 [s, 2H, RuC(Me)OCH2–]. 31P{1H} NMR (121 MHz, C6D6, 20°C):
δ 57.9 (s). 13C{1H} NMR (101 MHz, C6D6, 20°C): δ 19.7 [s, P(CHMe2)3], 20.5 [s,
P(CHMe2)3], 26.3 [vt, JP-C=9.7 Hz, P(CHMe2)3], 41.3 [s, RuC(Me)OCH2–],
71.3 [s, RuC(Me)OCH2–], 288.8 (t, 2JP-C=9.1 Hz, RuC(Me)OCH2–].CH(OCH2-CH2F).. Under
Ar, 1.85 g (11.5 mmol) of diethylamino sulfur
trifluoride (DAST)29 was dissolved in 10 mL CH2Cl2. The
solution was cooled to −78°C and 1.03 mL (11.5 mmol) of ethylene glycol vinyl ether was added via syringe. The solution was allowed to warm to room temperature and stirred for 1 h, then cooled to 0°C and the solvent removed to an N2 trap. The product was then fractionally distilled, collecting the 79–81°C
fraction (760 torr). Yield : approx. 500 mg (49%). 1H NMR (300 MHz, C6D6, 20°C):δ 3.28 (dt, 3JF-H=28.5 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F), 3.89 [dd, 3JH-H=6.9 Hz, 2JH-H=1.5 Hz, 1H, CH2CH(OR)], 4.01 [dd, 3JH-H=14.1 Hz, 2JH-H=1.5 Hz, 1H, CH2CH(OR)], 4.06 (dt, 3JF-H=47.7 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F), 6.30
[dd, 3JH-H=14.1 Hz, 3JH-H=6.9 Hz, 1H, CH2CH(OR)]. 13C{1H} NMR
(75.5 MHz, C6D6, 20°C): δ 67.0 (d, 2JF-C=20.5 Hz, OCH2CH2F),
81.5 (d, 1JF-C=171 Hz, OCH2CH2F), 86.1 [s, CH2CH(OR)], 151.7 [s,
CH2CH(OR)]. 19F NMR (282 MHz, C6D6, 20°C): δ
−224.8 (tt, 2JH-F=47.7
Hz, 3JH-F=28.5 Hz).C(Me)OCH2CH2F).. Under Ar, 10 mg
(0.022 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 2.5 μL (0.022 mmol) of 2-fluoroethyl vinyl ether
was added and the NMR tube sealed. 1H, 19F, and 31P{1H}
NMR spectra taken after 3 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2F). 1H NMR (300 MHz, C6D6, 20°C): δ
−21.32 (t, 2JP-H=21.6 Hz, 1H, RuH), 1.12 [dvt, JP-H=3JH-H=6.3
Hz, 18H, P(CHMe2)3], 1.25 [dvt, JP-H=3JH-H=6.3 Hz, 18H, P(CHMe2)3], 2.32–2.44 [m, 6H, P(CHMe2)3], 2.72 [s, 3H, RuC(Me)OR], 4.26 (dt, 2JF-H=47.7 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F), 4.46 (dt, 3JF-H=29.7 Hz, 3JH-H=3.9 Hz, 2H, OCH2CH2F). 31P{1H} NMR (121 MHz, C6D6, 20°C): δ 57.8 (s). 19F NMR (282 MHz, C6D6, 20°C): δ
−224.1 (tt, 2JH-F=47.7 Hz, 3JHF=29.7 Hz).C(Me)OCH2CH2NEt2).. Under Ar, 10 mg
(0.022 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 4.1 μL (0.022 mmol) of 2-(diethylamino)ethanol vinyl ether was added and the tube sealed. 1H and 31P{1H}
NMR spectra taken after 90 min revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CH2NEt2). 1H NMR (400 MHz, C6D6, 20°C): δ
−21.41 (brs, 1H, RuH), 0.96 [t, 3JH-H=7.2 Hz,
3H, N(CH2CH3)2], 1.16 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.29 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 2.40–2.50 (m,
6H, P(CHMe2)3], 2.47 [q, 3JH-H=7.2 Hz, 2H, N(CH2CH3)2], 2.74 [s, 3H, RuC(Me)OR], 2.77 (t, 3JH-H=6.8 Hz, 2H, OCH2CH2NEt2), 4.54 (t, 3JH-H=6.8 Hz, 2H, OCH2CH2NEt2). 31P{1H} NMR (162 MHz, C6D6, 20°C): δ 56.4 (s).C(Me)OCH2CHOCH2).. Under Ar, 10 mg
(0.022 mmol) of RuHCl(PPr3i)2 was placed in an NMR tube in
C6D6. Via syringe, 2.2 μL (0.022 mmol) of glycidyl vinyl ether (racemic) was added and the NMR tube sealed. 1H and 31P{1H} NMR spectra taken after 1 h revealed quantitative conversion to RuHCl(PPr3i)2(C(Me)OCH2CHOCH2). 1H NMR (300 MHz,
C6D6, 20°C): δ
−21.41 (t, 2JP-H=22.2 Hz, 1H, RuH), 1.15 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.26 [dvt, JP-H=3JH-H=6.4
Hz, 18H, P(CHMe2)3], 2.22 (dd, JH-H=5.3, 2.3
Hz, 1H, OCH2CHOCH2), 2.30 (apparent t, JH-H=4.4
Hz, 1H, OCH2CHOCH2), 2.35–2.45 [m, 6H, P(CHMe2)3],
2.72 [s, 3H, RuC(Me)OR], 3.00 (m, 1H, OCH2CHOCH2), 4.34
(dd, JH-H=13.4, 6.3 Hz, 1H, OCH2CHOCH2), 4.58 (dd, JH-H=13.4,
2.1 Hz, 1H, OCH2CHOCH2). 31P{1H} NMR (121 MHz,
C6D6, 20°C): δ 56.33, 56.24 (AB pattern, 2JP-P=220 Hz).HCl(PPr3i)2(C(Me)N(Me)C(O)CH3).. Under
Ar, 75 mg
(0.164 mmol) of RuHCl(PPr3i)2 was charged in a Schlenk flask
and dissolved in 10 mL of benzene. Via syringe, 18.6 μL (0.180 mmol) of N-methyl-N-vinylacetamide was added and the solution stirred for 4 days. The benzene was removed to a N2 trap and the purple solid was washed with pentane. Drying in
vacuo yielded 40 mg (45%) of RuHCl(PPr3i)2(C(Me)N(Me)C(O![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) )CH3).
1H NMR
(400 MHz, CD2Cl2, 20°C): δ
−17.24 (t, 2JP-H=26.2 Hz, 1H, RuH), 0.96
[dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.28 [dvt, JP-H=3JH-H=6.4
Hz, 18H, P(CHMe2)3], 2.20 [s, 3H, RuC(Me)NR2],
2.22–2.36 [m, 6H, P(CHMe2)3], 2.43 [s, 3H, N(Me)C(O)Me], 3.34 [s, 3H,
N(Me)C(O)Me]. 31P{1H} NMR (162 MHz, CD2Cl2, 20°C): δ 50.4
(s). 13C{1H} NMR (101 MHz, CD2Cl2, −20°C): δ 18.7 [s, P(CHMe2)3],
19.4 [s, P(CHMe2)3], 22.9 [s, N(Me)C(O)Me], 25.9 [vt, JP-C=8.6Hz, P(CHMe2)3], 34.7 [s, N(Me)C(O)Me], 40.9 [s, RuC(Me)NR2], 174.7 [s, N(Me)C(O)Me], 265.3 (t, 2JP-C=9.7 Hz, RuC). νCO(CH2Cl2, 25°C)=1599 cm−1.C(Me)NC(O)CH2CH2CH2).. Under Ar,
240 mg (0.524 mmol) of RuHCl(PPr3i)2
was charged in a
Schlenk flask and dissolved in 35 mL of benzene. Via syringe,
60.0 μL (0.561 mmol) of 1-vinyl-2-pyrrolidinone was added and the
solution stirred for 2 days. The benzene was removed to a liquid
N2 trap and the purple solid was washed
with pentane. Drying in vacuo yielded 160 mg (90% by 31P{1H} integration before workup) of RuHCl(PPr3i)2(C(Me)NC(O)CH2CH2CH2). 1H NMR
(400 MHz, CD2Cl2, 20°C): δ
−19.56 (t, 2JP-H=25.8 Hz, 1H, RuH),
0.98 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.27 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 2.01 [s, 3H, RuC(Me)NR2],
2.23–2.37 [m, 6H, P(CHMe2)3], 2.32 (apparent quintet, 3JH-H=7.5
Hz, 2H, NC(O)CH2CH2CH2), 2.52 [t, 3JH-H=8.0 Hz, 2H, NC(O)CH2CH2CH2],
3.70 [t, 3JH-H=7.0 Hz, 2H, NC(O)CH2CH2CH2]. 31P{1H} NMR (162 MHz, CD2Cl2, 20°C): δ 49.3 (s).
13C{1H} NMR (101 MHz, CD2Cl2, −20°C): δ 19.0 [s, P(CHMe2)3], 19.6 [s, P(CHMe2)3], 22.1 [s, NC(O)CH2CH2CH2], 25.9 [vt, JP-C=8.6 Hz, P(CHMe2)3], 29.7 [s, NC(O)CH2CH2CH2], 40.4 [s, RuC(Me)NR2],
45.9 [s, NC(O)CH2CH2CH2], 180.2 [s, NC(O)CH2CH2CH2], 261.5 (t, 2JP-C=10.0 Hz, RuC). νCO(CH2Cl2, 25°C)=1640 cm−1.CH–NC8H4O2).. Under
Ar, 100 mg
(0.218 mmol) of RuHCl(PPr3i)2 and 38 mg (0.218 mmol) of
vinyl phthalimide were mixed and then stirred 1.5 h at room
temperature. The solvent was removed and the product dried
in vacuo. Yield: quantitative. 1H NMR (400 MHz, THF-d8, 0°C): δ
−20.83 (dd, 2JP-H=15.6, 27.6 Hz, 1H, RuH), 1.10 [dvt, JP-H=3JH-H=7.6 Hz, 9H, P(CHMe2)3], 1.25 [m, 29H, P(CHMe2)3], 1.64 (brd, 1H, JH-H=3.2 Hz, CH2CH–NC8H4O2), 2.41 (dd, 1H, JH-H=14.0, 4.4 Hz, CH2CH–NC8H4O2), 2.74 [br s, 3H,
P(CHMe2)3], 2.85 [brs, 3H, P(CHMe2)3], 4.74 (ddd – apparent dt,
1H, JH-H=14.6, 6.4 Hz, CH2CH–NC8H4O2), 7.82 (m, 4H, CH2CH–NC8H4O2). 31P{1H} NMR (162 MHz, THF-d8, 0°C): δ 35.4,
57.9 (AX pattern, JP-P=278 Hz). 13C{1H} NMR (101 MHz,
THF-d8, −20°C): δ 19.5, 19.8, 20.16, 20.22 [s, P(CHMe2)3], 24.1
[d, JP-C=11.1 Hz, P(CHMe2)3], 24.8 [d, JP-C=16.1 Hz, P(CHMe2)3], 29.8
(s, CH2CH–NC8H4O2), 60.2 (s, CH2CH–NC8H4O2), 124.1, 124.7,
129.1, 130.8, 135.4 (s, CH2CH–NC8H4O2 aromatics; the peak
at 135.4 is over twice as large as the others and arises from
two signals with coincidental overlap), 167.0, 176.6 (s, CH2CH–NC8H4O2
carbonyls).
)CH3).
1H NMR
(400 MHz, CD2Cl2, 20°C): δ
−17.24 (t, 2JP-H=26.2 Hz, 1H, RuH), 0.96
[dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.28 [dvt, JP-H=3JH-H=6.4
Hz, 18H, P(CHMe2)3], 2.20 [s, 3H, RuC(Me)NR2],
2.22–2.36 [m, 6H, P(CHMe2)3], 2.43 [s, 3H, N(Me)C(O)Me], 3.34 [s, 3H,
N(Me)C(O)Me]. 31P{1H} NMR (162 MHz, CD2Cl2, 20°C): δ 50.4
(s). 13C{1H} NMR (101 MHz, CD2Cl2, −20°C): δ 18.7 [s, P(CHMe2)3],
19.4 [s, P(CHMe2)3], 22.9 [s, N(Me)C(O)Me], 25.9 [vt, JP-C=8.6Hz, P(CHMe2)3], 34.7 [s, N(Me)C(O)Me], 40.9 [s, RuC(Me)NR2], 174.7 [s, N(Me)C(O)Me], 265.3 (t, 2JP-C=9.7 Hz, RuC). νCO(CH2Cl2, 25°C)=1599 cm−1.C(Me)NC(O)CH2CH2CH2).. Under Ar,
240 mg (0.524 mmol) of RuHCl(PPr3i)2
was charged in a
Schlenk flask and dissolved in 35 mL of benzene. Via syringe,
60.0 μL (0.561 mmol) of 1-vinyl-2-pyrrolidinone was added and the
solution stirred for 2 days. The benzene was removed to a liquid
N2 trap and the purple solid was washed
with pentane. Drying in vacuo yielded 160 mg (90% by 31P{1H} integration before workup) of RuHCl(PPr3i)2(C(Me)NC(O)CH2CH2CH2). 1H NMR
(400 MHz, CD2Cl2, 20°C): δ
−19.56 (t, 2JP-H=25.8 Hz, 1H, RuH),
0.98 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 1.27 [dvt, JP-H=3JH-H=6.4 Hz, 18H, P(CHMe2)3], 2.01 [s, 3H, RuC(Me)NR2],
2.23–2.37 [m, 6H, P(CHMe2)3], 2.32 (apparent quintet, 3JH-H=7.5
Hz, 2H, NC(O)CH2CH2CH2), 2.52 [t, 3JH-H=8.0 Hz, 2H, NC(O)CH2CH2CH2],
3.70 [t, 3JH-H=7.0 Hz, 2H, NC(O)CH2CH2CH2]. 31P{1H} NMR (162 MHz, CD2Cl2, 20°C): δ 49.3 (s).
13C{1H} NMR (101 MHz, CD2Cl2, −20°C): δ 19.0 [s, P(CHMe2)3], 19.6 [s, P(CHMe2)3], 22.1 [s, NC(O)CH2CH2CH2], 25.9 [vt, JP-C=8.6 Hz, P(CHMe2)3], 29.7 [s, NC(O)CH2CH2CH2], 40.4 [s, RuC(Me)NR2],
45.9 [s, NC(O)CH2CH2CH2], 180.2 [s, NC(O)CH2CH2CH2], 261.5 (t, 2JP-C=10.0 Hz, RuC). νCO(CH2Cl2, 25°C)=1640 cm−1.CH–NC8H4O2).. Under
Ar, 100 mg
(0.218 mmol) of RuHCl(PPr3i)2 and 38 mg (0.218 mmol) of
vinyl phthalimide were mixed and then stirred 1.5 h at room
temperature. The solvent was removed and the product dried
in vacuo. Yield: quantitative. 1H NMR (400 MHz, THF-d8, 0°C): δ
−20.83 (dd, 2JP-H=15.6, 27.6 Hz, 1H, RuH), 1.10 [dvt, JP-H=3JH-H=7.6 Hz, 9H, P(CHMe2)3], 1.25 [m, 29H, P(CHMe2)3], 1.64 (brd, 1H, JH-H=3.2 Hz, CH2CH–NC8H4O2), 2.41 (dd, 1H, JH-H=14.0, 4.4 Hz, CH2CH–NC8H4O2), 2.74 [br s, 3H,
P(CHMe2)3], 2.85 [brs, 3H, P(CHMe2)3], 4.74 (ddd – apparent dt,
1H, JH-H=14.6, 6.4 Hz, CH2CH–NC8H4O2), 7.82 (m, 4H, CH2CH–NC8H4O2). 31P{1H} NMR (162 MHz, THF-d8, 0°C): δ 35.4,
57.9 (AX pattern, JP-P=278 Hz). 13C{1H} NMR (101 MHz,
THF-d8, −20°C): δ 19.5, 19.8, 20.16, 20.22 [s, P(CHMe2)3], 24.1
[d, JP-C=11.1 Hz, P(CHMe2)3], 24.8 [d, JP-C=16.1 Hz, P(CHMe2)3], 29.8
(s, CH2CH–NC8H4O2), 60.2 (s, CH2CH–NC8H4O2), 124.1, 124.7,
129.1, 130.8, 135.4 (s, CH2CH–NC8H4O2 aromatics; the peak
at 135.4 is over twice as large as the others and arises from
two signals with coincidental overlap), 167.0, 176.6 (s, CH2CH–NC8H4O2
carbonyls).Addition of 10 equiv. of the conjugated diene to a solution of 10 mg RuHCl(PPr3i)2 in 0.5 mL of C6D6 results in complete isomerization to 3-methylfuran within 10 min. No reaction occurs from the 3-methylfuran with RuHCl(PPr3i)2 at room temperature over several hours.
CD(OEt).. The isotopically labeled
olefin was prepared by hydrolysis of CH2C(Li)OEt31 with an excess of D2O in o-xylene at −10°C. Filtration and fractional distillation (30–35°C fraction) of the resulting mixture yielded 99% enriched CH2C(D)OEt by 1H NMR.Method 2 : RuDCl(PPr3i)2 was prepared by stirring RuHCl(PPr3i)2 in a small amount of acetone-d6 for 12 h at 25°C to allow isotopic exchange through the enol tautomer of the acetone-d6. Solution NMR shows >90% of the D incorporation now located in the PPr3i methyl groups. This is consistent with agostic stabilization of RuHClL2 by the PPr3i ligand(s).
X-Ray structure determinations
CCDC reference number 440/154. See http://www.rsc.org/suppdata/nj/a9/a907624G/ for crystallographic files in .cif format.
Acknowledgements
This work was supported by the U.S. NSF, the Université de Montpellier and the French CNRS.References
- J. N. Coalter, G. J. Spivak, H. Gérard, E. Clot, E. R. Davidson, O. Eisenstein and K. G. Caulton, J. Am. Chem. Soc., 1998, 120, 9388 CrossRef.
- D. Huang, G. J. Spivak and K. G. Caulton, New J. Chem., 1998, 22, 1023 RSC.
- J. N. Coalter, J. C. Huffman, W. E. Streib and K. G. Caulton, manuscript in preparation..
- We propose that the 1H chemical shift for R=Cy, SiMe3, and
CH2CH2CH2 (cyclic) are slightly downfield from that of R=Et as a result of the influence on the hydride of changing angle Cl–RuC due to electronic and steric factors with the bulkier R groups..
- Remarkably, 2,3-dihydropyran, the six-membered ring analog, fails to react after 12 h at 25°C..
- (a) P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100; Search PubMed; (b) S. T. Nguyen, L. K. Johnson, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1992, 114, 3974; Search PubMed; (c) Z. Wu, S. T. Nguyen, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1995, 117, 5503; Search PubMed; (d) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039. Search PubMed.
- Slower reaction rate with H2CCD(OEt) permits
observation of this AB NMR pattern even at 25°C..
-
Even
if H2CCD(OEt) is reacted with RuHClL2 (2:1 mole ratio) at 25°C to
minimize the time spent as the olefin
adduct, D is found statistically in RuD, RuCC(H,D)3(OEt) and
all three vinylic positions of free ethyl vinyl ether; this procedure
only serves to decrease the deuteration in the phosphine
methyl groups..
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1. Search PubMed.
- F. Ford, T. Yuzawa, M. S. Platz, S. Matzinger and M. Fülscher, J. Am. Chem. Soc., 1998, 120, 4430 CrossRef CAS.
- T. J. Johnson, A. Albinati, T. F. Koetzle, O. Eisenstein, J. C. Huffman and K. G. Caulton, Inorg. Chem., 1994, 33, 4966 CrossRef CAS.
- M. Olivàn, E. Clot, O. Eisenstein and K. G. Caulton, Organometallics, 1998, 17, 3091 CrossRef CAS.
- M. M. Gallo, T. P. Hamilton and H. F. Schaefer, J. Am. Chem. Soc., 1990, 112, 8714 CrossRef CAS.
- J. Silvestre and R. Hoffmann, Helv. Chim. Acta, 1985, 68, 1461 CrossRef CAS; Y. Wakatsuki, N. Koga, H. Werner and K. Morokuma, J. Am. Chem. Soc., 1997, 119, 360 CrossRef CAS.
- S. F. Vyboishchikov and G. Frenking, Chem. Eur. J., 1998, 4, 1428 CrossRef CAS.
- A. F. Hill, W. R. Roper, J. M. Waters and A. H. Wright, J. Am. Chem. Soc., 1983, 105, 5939 CrossRef CAS.
- C. P. Casey, C. J. Czerwinski, D. P. Powell and R. K. Hayashi, J. Am. Chem. Soc., 1997, 119, 5750 CrossRef CAS.
- (a) A. Cutler, S. Raghu and M. Rosenblum, J. Organomet. Chem., 1974, 77, 371; Search PubMed; (b) T. C. T. Chang, M. Rosenblum and S. B. Samuels, J. Am. Chem. Soc., 1980, 102, 5931; Search PubMed; (c) H. Chen and W. D. Harman, J. Am. Chem. Soc., 1996, 118, 5672. Search PubMed.
- W. D. Harman, Chem. Rev., 1997, 97, 1953 CrossRef CAS.
- J. S. Freundlich, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1996, 118, 3643 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head–Gordon, C. Gonzalez and J. A. Pople, GAUSSIAN 94, Revision B3, Gaussian Inc., Pittsburgh, 1995. Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. G. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- A. H. Höllwarth, M. B. Böhme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 237 CrossRef.
- P. C. Harihan and J. A. Pople, Theor. Chem. Acta, 1973, 28, 213 Search PubMed.
- C. Grünwald, O. Gevert, J. Wolf, P. González-Herrero and H. Werner, Organometallics, 1996, 15, 1960 CrossRef CAS.
- T. E. Wilhelm, T. R. Belderrain, S. N. Brown and R. H. Grubbs, Organometallics, 1997, 16, 3867 CrossRef CAS.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS.
- W. Miles, C. Berreth and P. Smiley, Tetrahedron Lett., 1993, 34, 5221 CrossRef CAS.
- R. Knorr and T. Roman, Angew. Chem., Int. Ed. Engl., 1984, 23, 366 CrossRef.
Footnote |
| † Non-SI units employed: 1 torr≈133 Pa; 1 kcal≈4.18 kJ. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2000 |