Coupling of protein sheet crystals (S-layers) to phospholipid monolayers†
Markus Weyganda, Manfred Schalkea, Paul B. Howes‡b, Kristian Kjaerb, Jaqueline Friedmannc, Barbara Wetzer§c, Dietmar Pumc, Uwe B. Sleytrc and Mathias Löschea
aLeipzig University, Institute of Experimental Physics I, D–04103, Leipzig, Germany. E-mail: loesche@physik.uni-leipzig.de
bRisø National Laboratory, Department of Solid State Physics and Chemistry, DK–4000, Roskilde, Denmark
cUniversity for Agricultural Sciences, Center for Ultrastructure Research and Ludwig-Boltzmann-Institute for Molecular Nanotechnology, A–1180, Vienna, Austria
First published on UnassignedUnassigned22nd December 1999
Abstract
The coupling of bacterial S-layer proteins to phospholipid membranes has been studied in molecular detail with respect to, particularly, the lipid headgroups. Emphasis has been laid on two of the best characterized protein species, the S-layer protein from Bacillus sphaericus CCM2177 and from Bacillus coagulans E38–66/V1. A combination of fluorescence microscopy, surface sensitive scattering techniques (grazing-incidence X-ray diffraction as well as X-ray and neutron reflectometry) and infrared spectroscopy (FT-IRRAS), applied to surface monolayers of lipids onto which the protein has been reconstituted as continuous molecular crystal sheets, provides a wealth of information which has been utilized to propose detailed molecular models.
Introduction
Protein–lipid interactions play an important role in life science. In biomimetic approaches to materials science, well-defined proteinaceous interface layers gain progressively more importance in various fields, such as biosensorics,1 biocatalysis,2 and the build-up of well-defined supramolecular architectures at interfaces.3,4 Since a large variety of biological processes is membrane mediated, there is great interest in the meso- and macroscopic reconstitution of functional lipid membranes.5 On the other hand, the use of free-standing membranes is primarily impeded by a low stability. It was recently demonstrated that the stability of such membranes can be significantly increased by the recrystallization of isolated S-layer proteins. Such composite S-layer–lipid films are biomimetic structures which resemble those archaeal cell envelopes that are exclusively composed of monomolecular arrays of (glyco)proteins and a closely associated plasma membrane.6 Bacterial and archaeal surface layers (S-layers) are monomolecular protein sheet crystals covering the outer surface of prokaryotic organisms.7,8 They constitute the outermost component of such cells providing mechanical support to the cell in archaeae and control material transfer from and into the cell as their crystal lattice incorporates pores. Most S-layers consist of one single protein (or glycoprotein) species that is specific for the organism it derives from. While S-layer proteins reassemble into sheet crystals under a variety of experimental conditions, three-dimensional crystallization has not been achieved to date for any protein of this class so that atomic level structural information is not at hand. On the molecular scale, the topographical properties of S-layers and in particular the topography of the S-layer lattice from Bacillus coagulans E38–66/V1 have been revealed by electron microscopy.9 The structural properties of the S-layers from various organisms may vary greatly7,10 (Mr between ~40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and ~200000; lattice constants between ~3 and ~30 nm; S-layer thickness between ~5 and ~15 nm). Other features are quite similar for many of the S-layers studied so far: Protein within the S-layer occupies only a fraction of the area, typically 30–70%, such that large water-filled pores span the S-layer. A pronounced asymmetry of the topographical and physico-chemical properties of the two faces oriented to the cell wall (“inner face") or pointing away to the environment (“outer face") has been observed for various S-layer species: The inner surfaces are generally more corrugated than the outer ones and bear net negative charges.11 The overall amino acid composition is similar for many species studied to date, with a high abundance of glutamic and aspartic acid and little or no sulfur-containing amino acids.7,12 In Gram-positive bacteria, such as the ones studied in this paper, the S-layer is associated with the peptidoglycane-containing cell wall, but not with a cytoplasmatic membrane as in most archaeae.
000 and ~200000; lattice constants between ~3 and ~30 nm; S-layer thickness between ~5 and ~15 nm). Other features are quite similar for many of the S-layers studied so far: Protein within the S-layer occupies only a fraction of the area, typically 30–70%, such that large water-filled pores span the S-layer. A pronounced asymmetry of the topographical and physico-chemical properties of the two faces oriented to the cell wall (“inner face") or pointing away to the environment (“outer face") has been observed for various S-layer species: The inner surfaces are generally more corrugated than the outer ones and bear net negative charges.11 The overall amino acid composition is similar for many species studied to date, with a high abundance of glutamic and aspartic acid and little or no sulfur-containing amino acids.7,12 In Gram-positive bacteria, such as the ones studied in this paper, the S-layer is associated with the peptidoglycane-containing cell wall, but not with a cytoplasmatic membrane as in most archaeae.While they are in vivo attached to cell walls, S-layer proteins isolated from B. coagulans E38–66/V1 and B. sphaericus CCM2177 are rather easy to recrystallize at a large variety of interfaces—including lipid surfaces.13 With the goal to use such reconstituted S-layers for the stabilization of model membranes, we have investigated the—unnatural—association and recrystallization of these two protein species at phospholipids. Recrystallized S-layers, particularly of the protein derived from B. coagulans E38–66, may cover holes several microns large, and larger areas of phospholipid films have been observed to be overgrown by molecularly thin polycrystal films.14–16 We have demonstrated that the attachment of monomolecular protein sheet crystals stabilize the fragile lipid double layer in membrane models used for biophysical studies.17,18 Since S-layers are natural molecular sieves with a well-defined porosity, such an attached protein sheet crystal does not inhibit molecular diffusion processes to the membrane and allows for manipulation of the membrane model. In order to understand the interaction and the coupling between protein and lipid in molecular detail, we have set out to investigate such molecular hetero-layer systems with a variety of surface sensitive characterization techniques including fluorescence microscopy, X-ray and neutron scattering, and FTIR spectroscopy.
Materials and methods
B. sphaericus strain CCM2177 was obtained from the Czech Collection of Microorganisms (Brno, Czech Republic). B. coagulans E38–66/V1 was from F. Hollaus (Zuckerforschung Tulln GmbH, Tulln, Austria). Growth conditions of the bacteria in continuous culture were as reported,19 and extraction of the S-layer protein with guanidine hydrochloride (GHCl, 5 M in 50 mM Tris-HCl buffer, pH 7.2, 20°C) was performed as described.19 GHCl extracts were dialyzed against H2O (B. sphaericus) or 10 mM CaCl2 (B. coagulans). Dipalmitoylphosphatidylethanolamine (DPPE), dimyristoylphosphatidylethanolamine (DMPE) and dipalmitoylphosphatidylcholine (DPPC) were purchased from Avanti Polar Lipids, Inc. (Birmingham, AL) and used as received. Lipids were dissolved in CHCl3–CH3OH (3∶1, Merck, Darmstadt, Germany, pro analysi grade) to form a spreading solution of ∼1.0 mg ml−1. Ultrapure H2O was prepared by filtering in a Milli-Q (Millipore, Bedford, MA) apparatus. For a translation of the scattering length density profiles obtained in reflectivity experiments (see below) into mass density, the amino acid composition of the proteins has been determined.12All experiments were performed at room temperature (T = 21 ± 1°C). Self-assembly products of the purified S-layer proteins were sedimented at 40000 g and T = 4°C immediately before use. The clear supernatant containing protein monomers at a concentration of typically 2 mg ml−1 was injected underneath phospholipid monolayers preformed at the desired lateral pressure π. In typical experiments, π was observed to increase slightly after protein injection, e.g. by a few mN m−1.16 The protein was allowed to recrystallize over night for most of the experiments reported here.
Fluorescence microscopy for the observation of molecular protein/lipid layer systems at aqueous surfaces has been described in detail.20 For dual label experiments, the S-layer protein from B. coagulans was labeled with carboxyfluorescein (CFS) as described20 and was observed in conjunction with phospholipid surface monolayers that contained ∼2 mol% of a sulforhodamin-(SR-) labeled DPPE. By means of the different labels, structure formation in the protein and lipid layers may be well discriminated.
X-Ray reflectivity and diffraction (grazing-incidence X-ray diffraction, GIXD) experiments were performed at the BW1 beam line of HASYLAB (DESY, Hamburg, Germany).21 In addition, X-ray reflection measurements in a more confined momentum transfer regime were taken on a laboratory scale instrument in Leipzig.22 Neutron reflectometry was done at the new Mark II instrument, equipped with Langmuir-type liquid surface sample cell, at port TAS9 in the guide hall of the DR3 reactor at Risø National Laboratory (Roskilde, Denmark). All surface scattering experiments were performed using home-built Langmuir film balances (surface area 16 × 30 cm2) incorporated in gas-tight, thermostated Al containers with Kapton (X-ray) or Al (neutron experiments) windows for the beam.23 Polished (λ/10) Pyrex (boron silicate) glass blocks, inserted into the subphases to diminish the depth under the beam footprint on the monolayer to ∼300 mm, were used to suppress surface waves in the film balances.
Reflectivity data were interpreted in terms of a model-free constrained least-squares approach introduced by Skov Pedersen and Hamley24–26 where the scattering length density distribution ρ(z) across the interface is described by B-spline functions [eqn. (1)].
 | (1) |
As a function of the horizontal component Qxy ∼ (4π/λ)sin(2θxy/2) of the scattering vector, the intensity diffracted from 2D crystal patches in a lipid surface monolayer peaks at Bragg positions Qxyhk. In the absence of a crystalline repeat in the vertical (z) direction, the intensity extends as a smooth function, the Bragg rod profile Ihk(Qz), of the vertical component of the scattering vector, Qz∼(2π/λ)sin(αf), where αf is the vertical direction of the scattered rays. The Bragg rod profiles resulting from the phospholipid chains may be modeled27,28 by eqn. (2),
 | (2) |
 | (3) |
Infrared reflection-absorption spectroscopy (FT-IRRAS) measurements31,32 were performed with a Bio-Rad (Digilab) FTS 60A spectrometer equipped with an MCT detector. The monolayer was spread in a home-made thermostated Langmuir trough that is mounted on a shuttle device together with another trough containing pure water33 which allows the IR beam to be switched between the sample and a reference area. Spectra were acquired by coaddition of 1024 scans with a resolution of 8 cm−1.
Motivation
We have studied the structure formation in lipid monolayer–recrystallized S-layer systems on the mesoscopic length scale with fluorescence microscopy (FM) and electron microscopy (EM). In recent extensive EM investigations, we addressed systematically the question of which lipids and which experimental conditions support the recrystallization of the protein from B. coagulans E38–66/V1 into well-ordered, coherent S-layers.16 In older FM and FTIR studies, we have investigated the interplay between order and disorder in the system.20 In that study, we used a dual label technique to discriminate between structure formation in a phospholipid (DMPE) monolayer, labeled with small amounts of SR-DPPE and adsorption and recrystallization of CFS-labeled protein. When the phospholipid monolayer, which served as a crystallization matrix for the proteins, was prepared in a phase separated (liquid expanded/liquid condensed, LE/LC) state,¶ it was observed that the protein adsorbed preferentially to those areas in the monolayer that were of lower molecular order with respect to the lipid acyl chains. However, nucleation of protein crystals occurred inevitably at locations on the phase boundaries (cf.Fig. 1 a/b of ref. 20). Subsequently, crystallization was only observed to occur underneath the ordered lipid phase (cf.Fig. 1 e/f of ref. 20). Only after long incubation times did we observe S-layers that had overgrown the entire surface area, i.e., protein crystals that were attached to areas covered by both ordered and disordered lipid. On the other hand, FTIR spectra of the acyl methylene vibrations indicated an increase in the (average) order parameter, which suggests that the association of protein with disordered lipid has been driving the lipid chains into a state of higher order.20 The complex interplay between order and disorder in this system is schematically depicted in Fig. 1. From indirect arguments it was postulated that the protein, upon attachment to the lipid-covered interface, is unlikely to interpenetrate into the alkane phase. Rather, it was argued, the control of the protein crystallization process, which requires information transfer from the lipid to the interface, occurs via the lipid headgroups, and it was hence concluded that the protein is likely to interpenetrate the headgroups—but not the acyl chains—of the lipid monolayer.20 | ||
| Fig. 1 Schematic representation of the adsorption and recrystallization of S-layer protein monomers from Gram-positive bacteria at phospholipid monolayers as suggested from fluorescence microscopic studies. (a) Protein monomers injected into the subphase underneath a monolayer that has been prepared in a phase separated (LE/LC) state adsorb preferentially to the more disordered LE phase. (b) Nucleation of protein sheet crystals occurs at the LE/LC phase boundary and (c) protein recrystallization proceeds specifically underneath the ordered lipid phase, LC. (d) Only after long incubation times, the reconstituted S-layer covers the entire surface. As evidenced from IR spectroscopy, protein association drives the lipid into a state of higher local order. | ||
In systematic recrystallization experiments, using EM on transferred and stained protein/lipid interface layers, we have addressed the question of which lipid species and which experimental conditions sustain the recrystallization of the S-protein from B. coagulans into reconstituted S-layers.16 It was generally observed that disordered lipid acyl chains completely inhibit protein recrystallization at pH >4. It was also found that the chemical identity of the lipid headgroups, as well as divalent cations in the subphase, significantly determine the propensity of the proteins for recrystallization:16 Anionic lipids on pure water do not support protein recrystallation at all. Underneath zwitterionic lipid headgroups, on the other hand, recrystallization of the protein leads to large S-layer lattices, particularly if Ca2+ is present in the subphase in mM quantities. Whereas recrystallization at PE interfaces requires Ca2+, conceivably to make the headgroups available for protein binding by interrupting the hydrogen-bond network that interlocks the PE groups, proteins recrystallize readily at PC interfaces with or without Ca2+.16 While Ca2+ induces protein recrystallization even underneath anionic lipids if their headgroups are small (phosphatidic acid, PA), steric repulsion of larger anionic groups (phosphatidylglycerol, PG) inhibits protein attachment to the interface under all experimental conditions. Under a variety of experimental conditions at pH≤4.3|| and also at clean water surfaces, protein binding and recrystallization is observed in an upside-down orientation, i.e., with the inner face pointing to the subphase compartment. This orientation is distinguished from the “natural" orientation by virtue of the handedness of the oblique S-layer lattice of this protein.14 It appears thus16 that protein binding is determined by anionic sidegroups on the inner surface of the protein, presumably buried in a rugged surface topology that imposes steric constraints on the binding process. Binding of cationic groups to these “primary" sites occurs in a specific manner in which different lipid headgroups are discriminated against each other. The binding occurs either to partial cationic charges, exposed on the outside region of the lipid headgroups, or to divalent cations which are presumably attached to the phosphate groups, which are located more in the center of the headgroup layer.
While these mesoscopic studies yielded a wealth of detailed information, it is important to remember that the molecular scale interpretation is rather indirect. We have thus undertaken a study of the S-layer protein interactions with phospholipid interfaces on the microscopic length scale. These investigations are in the focus of the current paper.
Molecular scale structure
Fig. 2 shows the Bragg reflections, measured in a GIXD experiment, from a quasi-two-dimensional (2D) S-layer lattice formed after incubation of a DPPE monolayer—prepared on an unbuffered subphase containing 10 mM CaCl2 and compressed to a lateral pressure π = 28 mN m−1—with the protein from B. coagulans E38–66/V1. It shows (panel a) a contour plot of the scattered intensity Ivs. the horizontal and vertical components, Qxy and Qz, of the momentum transfer and (panel b) the Qz-integrated intensity I vs. Qxy. The Bragg positions (solid vertical lines) are indexed assuming an oblique lattice with |a| = 99.5 Å, |b| = 76 Å, and γ = 80°. For clarity, third order positions (or higher) are shown as broken lines in Fig. 2b only and have not been labeled. The indexing scheme describes well the observed peak positions for Qxy> 0.1 Å−1. At lower Qxy, the coincidence appears worse because of the large sloping background that has not been corrected. The results are consistent with earlier TEM work in which an oblique lattice with |a| = 94 Å, |b| = 74 Å, and γ = 80° has been observed. Note that the diffraction pattern is quite distinct from the pattern observed with S-layers from B. sphaericus CCM2177, which was earlier studied intensively with surface sensitive scattering techniques.12 Whereas for S-layers from B. sphaericus the scattering intensity is located primarily in the {1,0} and {1,1} peaks in Qz positions off the horizon (Fig. 3 of ref. 12), in the case of the S-layer from B. coagulans the intensity is concentrated mainly at the horizon (Qz ∼ 0) and is more evenly distributed over the various {h,k} peaks.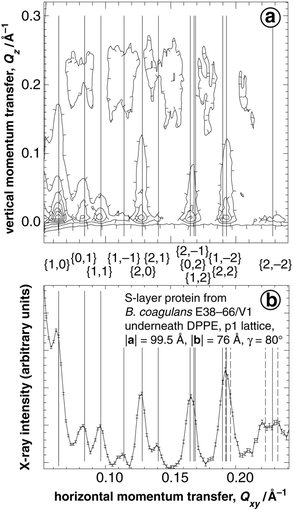 | ||
| Fig. 2 GIXD from an S-layer lattice (B. coagulans E38–66/V1) reconstituted underneath a DPPE monolayer. (a) Contour plot of the scattered intensity I vs. the horizontal and vertical components, Qxy and Qz, of the momentum transfer. Note that the intensity as a function of Qxy is concentrated in Bragg peaks which, vertically resolved, are seen to extend as Bragg rods in the Qz-direction. (b) The intensity integrated over Qz for −0.05 < Qz < 0.45 Å−1 is plotted as a function of Qxy. The vertical lines mark Qxy-positions of the Bragg maxima corresponding to the indicated {h,k} Miller indices of an oblique lattice with |a| = 99.5 Å, |b| = 76 Å and γ = 80°. Dashed vertical lines in (b) indicate third order Bragg positions that have not been labeled. | ||
 | ||
| Fig. 3 GIXD from a DPPE monolayer at before (a, b; π ∼ 28 mN m−1) and after (c, d; π ∼ 35 mN m−1) adsorption and crystallization of S-layer protein from B. coagulans E38–66/V1 on pure water. The surface area has been kept constant during the S-lattice formation. Top: contour plot of I(Qxy, Qz). Bottom: Qz-integrated diffraction intensity I(Qxy). Adapted from ref. 12. | ||
Independent of the reconstituted S-layer lattice, the order of the lipidic acyl chains may be characterized with GIXD by just tuning to the appropriate Qxy region.12Fig. 3 shows the diffraction pattern of a DPPE monolayer on 10 mM CaCl2 at π = 28 mN m−1 prior to protein injection (panel a) and after reconstitution of an S-layer from B. coagulans (panel c) and (panels b and d) the Qz-integrated intensities vs.Qxy. It is obvious that the phospholipid monolayer undergoes only minimal reorganization upon protein adsorption and recrystallization. A slight increase of the center position of the Bragg peak indicates a lateral compression of the acyl chains on the order of ΔA/A = 1%, where A is the area per acyl chain in the monolayer. The slight compression of the contour profile of the scattered intensity along Qz indicates a marginal reduction of the tilt angle α of the chains against the surface normal: a quantitative evaluation is consistent with a change from α ∼ 9° to 6°.
As might be expected in view of the remarkable differences between the GIXD patterns of the protein lattices from B. coagulans and B. sphaericus, the protein density distributions of the two S-layers along the z direction, as derived from X-ray reflectometry, differ quite drastically. Fig. 4 shows a reflectogram of an S-layer reconstituted at a DPPE monolayer on 10 mM CaCl2 in comparison with that of the DPPE monolayer (π = 28 mN m−1) prior to protein incubation. The solid lines are the calculated reflectivities corresponding to the electron density models shown in panel b. In earlier work12 we have shown that the electron density contribution, and hence the volume density, of the S-layer from B. sphaericus shows a bimodal distribution that peaks at z = −45 and −105 Å from the surface layer/air interface (corresponding to ∼−20 and −80 Å from the protein/lipid interface) and that the S-layer thickness is dCCM2177 ∼ 90 Å. This implies that a major part of the protein mass is located close to the outer surface of the S-layer for that bacterial species. In contrast, the data for the B. coagulans protein suggest that the center of mass is located close to the S-layer/lipid interface, at z ∼−40 Å from the surface layer/air interface. The outer surface of the S-layer, and thus its thickness, cannot be precisely determined for B. coagulans from X-ray reflectometry owing to the smooth decay of the electron density contribution of the reconstituted S-layer along the direction of the subphase. The differences in the protein mass density distributions of the two protein species are related to the differences of the diffraction patterns and may explain in qualitative terms why the intensities in the Bragg rods of the diffraction pattern specifically from the reconstituted B. coagulans S-layer are concentrated at the horizon.
 | ||
| Fig. 4 X-Ray reflectivity of a DPPE monolayer prior to and after the reconstitution of an S-layer (B. coagulans E38–66/V1). Panel (a) Data (open symbols) and models (solid lines) corresponding to the electron density profiles ρ(z) shown in (b). For clarity, the reflectivity curve of the pure DPPE monolayer (lower trace) has been multiplied by 10−2. In panel (b), ρ(z) of the pure DPPE monolayer is shown as a dashed line and that of the protein–lipid layer system as a solid line. | ||
Further detail on the interactions between S-layer protein and lipid monolayers is obtained from FT-IRRAS. Fig. 5 shows spectra of lipid headgroup vibrations in the range ν = 1000–1300 cm−1. At least four prominent peaks are identified in this spectral interval: the asymmetric and symmetric phosphate stretch vibrations, νas(–PO2−–) and νs(–PO2––), around 1225 and 1080 cm−1, respectively, as well as the carbonyl ester vibrations, νas(–CO–O–C–) and νs(–CO–O–C–), between 1165 and 1180 cm−1 and around 1030 cm−1. Shown in Fig. 5a are spectra from DPPE monolayers without protein on pure water and on 10 mM CaCl2 in comparison with a spectrum taken after the reconstitution of an S-layer from B. coagulans E38–66/V1. Fig. 5b displays spectra involving DPPC. The spectrum of the lipid monolayer without protein on pure water is essentially identical to that on CaCl2-containing subphase and has been omitted for clarity. A comparison of the three traces concerning the PE headgroups in Fig. 5a shows a successive shift of the phosphate stretch vibrations to higher energies in the sequence from the monolayer on water, on Ca2+-containing subphase and in the protein-bound state. In contrast, the PC phosphate vibrations seem not at all affected by the addition of Ca2+ to the subphase (data not shown) or by protein binding. On the other hand, a major change is observed in the νas(–CO–O–C–) vibration upon protein binding, with absorption shifted from the 1180 cm−1 range to about 1165 cm−1. In this spectral region, the situation is not clear in the spectra taken for the PE monolayers. The most conservative conclusion that may be drawn from these data is that with PE monolayers as a recrystallization matrix, the protein interpenetrates the lipid headgroups such that the phosphate groups are affected in their molecular interactions. With PC monolayers, peptide insertion may even affect the carbonyl esters at the boundary between the hydrophilic and the hydrophobic parts of the monomolecular lipid layer. More speculative suggestions, as to what the observed spectral changes could relate to, will be put forward in the next section.
 | ||
| Fig. 5 FTIR spectra in the region of the ester carbonyl and phosphate stretch vibrations of the phospholipid headgroups. | ||
Discussion
The non-natural association of S-layer proteins from the two Gram-positive Bacillus species B. coagulans E38–66/V1 and B. sphaericus CCM2177 with phospholipid membranes and their recrystallization into coherent S-layer lattices bears many similarities as far as the interface between the proteins in the crystal and the lipid headgroups are concerned. Major differences have been documented between the internal structures of the S-layers.The electron density profiles presented in Fig. 4 together with the GIXD data shown in Fig. 3 provide clear evidence that the structure of the lipid acyl chains is at best marginally affected by protein adsorption and recrystallization. In both cases studied the electron density distributions within the lipid headgroups are slightly altered. In a refined structural model of the S-protein/lipid headgroup interface within the layer system formed by the B. sphaericus CCM2177 protein at DPPE, derived from a simultaneous evaluation of the X-ray data with neutron reflectivity data of reconstituted S-layers under perdeuterated DPPE (DPPE-d62) on H2O and D2O, we have specifically investigated the distribution of protein material and water within the boundary region.35 We find that peptide interpenetrates deeply into the lipid headgroups—without affecting the lipid acyl chains. Concomitantly, water is introduced into the headgroup region. In view of an essentially unchanged area per lipid within the surface film, volume conservation requires that the headgroups tilt toward the surface normal to make space available for the peptide. Quantitatively, it is estimated that ∼65 electrons associated with peptide material insert into the lipid headgroup region per lipid molecule in the monolayer. Since the coherence area within the aqueous surface which is averaged in a typical reflectivity experiment is mesoscopic, micron-sized along the projection of the beam direction on the footprint,36 it is impossible to decide whether this material inserts homogeneously—on average one amino acid sidechain per lipid—or inhomogeneously—with larger peptide loops associating with only a few of the ∼100 DPPE molecules located within the area that one S-layer protein of the B. sphaericus CCM2177 variety occupies in the surface film. In view of the specificity of S-protein binding to lipids that has been revealed from systematic investigations of recrystallization conditions16 it seems quite clear that the lipid headgroups interact with spatially organized peptide structures, which suggests that the latter scenario is more realistic.
While we do not have such detailed information from (neutron) reflectivity measurements available on the interface between the B. coagulans S-layer adhering to phospholipid headgroups, it is quite reasonable to assume that the situation there is similar. This is corroborated by the IR results shown in Fig. 5 which demonstrate that vibrational modes of the phosphates are affected by the protein in PE headgroups and that within the PC headgroups even the carbonyl esters at the boundary between the hydrophilic and the hydrophobic parts of the lipids show changes that may be associated with peptide interpenetration into the monolayer.
The molecular origin of these spectral changes is currently still a matter of debate. Generally, the structure of the νas(–PO2––) has been related to the hydration state of phospholipid headgroups37,38 and both monolayer compression and the addition of Ca2+ has been implied to dehydrate PC headgroups as indicated by a shift of νas(–PO2––) to higher energy.39 In PE headgroups we clearly observe a shift in νas(–PO2––) from 1220 to 1225 cm−1 in the sequence (PE on water), (PE w/10 mM Ca2+), (PE w/Ca2+and associated S-layer). This shift is of the same magnitude as has been reported for (PC on water), (PC w/5 mM Ca2+).39 However, we do not observe any measurable shift in that spectral region with PC monolayers upon S-layer reconstitution (Fig. 5b). An alternative interpretation of the spectral effects in PE may thus relate the shifts to hydrogen bonding (–NH3+ –PO2––) within or between the PE headgroups that might be successively disrupted by Ca2+ and by the insertion of peptide into the headgroups. The latter effect—the disruption of hydrogen bonds due to the insertion of peptide—would be expected as a consequence of a collective tilting of the headgroups toward the surface normal as hydrogen bonds among adjacent PE's are only formed if the (–NH3+) and (–PO2––) moieties are located in the same plane parallel to the interface. In that context it has also been explained by the formation of hydrogen bonds between the headgroups and their disruption by Ca2+ that S-layer proteins do not bind and recrystallize at PE interfaces on pure water while they form well-ordered S-layers in the presence of the cation.16 The model would also explain why one does not observe a similar frequency shift of the νas(–PO2––) in PC as in PE upon S-layer reconstitution as PC is incapable of forming intramolecular H-bonds due to the lack of a proton donator group.
The spectral changes observed with DPPC upon S-layer protein reconstitution in the region between 1150 and 1200 cm−1 is associated with conformational changes of the glycerol backbone. The νas(–CO–O–C–), which gives rise to adsorption in this region, is centered around 1165 or 1180 cm−1, depending on whether the (–CO–O–C–) fragment is oriented more normal or more parallel to the interface.37 In lipid crystals40 as well as in model membranes,41 the linker fragment of the acyl chain at the β position is preferentially oriented along the surface normal, whereas at the α position it is oriented parallel before the chain turns and points away from the interface. This is presumably also true in the DPPC monolayer on Ca2+-containing subphase (trace 1 in Fig. 5b), where the spectral intensity is rather uniformly distributed between the 1165 and the 1180 cm−1 positions. In contrast, upon S-layer reconstitution we observe a redistribution of the absorption from 1180 to 1165 cm−1 (see arrows in Fig. 5b), which may be indicative of a conformational change of the linker fragment at the β position into an orientation more parallel to the interface. Whether this change is a direct or indirect consequence of peptide insertion into the headgroup is not clear; it should be noted, however, that similar spectral differences may also be observed between DPPC monolayers that have been compressed with greatly differing barrier speeds (M. Schalke, unpublished results). As the spectra in Fig. 5 have been obtained under comparable preparation conditions with respect to the lipid monolayer we infer from these results that the peptide material from the S-layer inserts deeply into the PC moiety, leading to a gross reorganization of the lipid headgroups.
Both GIXD and reflectometry reveal major differences in the internal structure of the S-layer between the two protein species. This is not too surprising since the two proteins have significantly different molecular weights (Mr
∼ 100000 and 120000 for the B. coagulans and the B. sphaericus proteins, respectively) and amino acid composition. Whereas the S-layer of B. sphaericus has a square lattice, that of B. coagulans has a lattice of oblique symmetry. In addition, the protein density distribution across the S-layer is characteristically different for the two proteins: While the B. sphaericus S-layer has two distinct density maxima with a pronounced minimum in between, and has a layer thickness in excess of 90 Å,12 the B. coagulans protein density distribution exhibits just one hump, which is located close to the lipid monolayer, and extends only ∼40 Å along the z direction. This indicates a lipid–protein layer structure, shown schematically in Fig. 6, which is quite different from that of the B. sphaericus S-layer (compare with Fig. 6 in ref. 12). Integration over the electron density distribution yields estimates of the partial volume of the protein and the number of electrons associated with the protein per unit area. Let ρlipid(z) be the electron density profile of the lipid-only system (dashed line in Fig. 4b) and ρ(z) the electron density profile of the lipid–protein system (full line in Fig. 4b). Then we may estimate the distribution of protein volume fraction ξpr(z) from eqn. (4),
 | (4) |
 | (5) |
000 Å3 (per unit cell size) and the number of electrons, nepr [eqn. (6)],  | (6) |
700 and VE38–66
∼ 118700 Å3. This estimate provides an important consistency check on the evaluation procedure and shows that one morphological unit is contained within the unit cell area, Aunit cell
∼ 7500 Å2. The latter result has been also derived from EM characterization of the S-layers.9 | ||
| Fig. 6 Schematic of the lipid–protein multilayer structure for the S-layer protein from B. coagulans E38–66/V1 as it emerges from the analysis of the electron density profile, indicated on the left-hand side. The appearance of the protein is consistent with the electron density profile, and proteins and lipids are approximately drawn to scale. There is, however, no structural information for the protein available to date on the atomic scale. | ||
Conclusions
The motivation for this paper has two major components: (a) to summarize the molecular details of the coupling of the S-layer protein from, specifically, B. coagulans E38–66/V1 to phospholipid monolayers and to compare these details with the data—published earlier12—for the protein from B. sphaericus CCM2177 and (b) to review the state of the art in surface sensitive structure characterization techniques at the air/water interface that have been used to reveal this information. As regards the former of these goals it has been shown that the molecular details of the coupling, at the level of the protein/lipid interface, are quite similar for the two protein species although the protein structure—and hence the S-layer structure—seem rather different. On the experimental side, the methods applied for the structural characterization of S-layers coupled to lipid surface monolayers are not restricted to large proteins, such as the S-layer proteins investigated, but are quite general: peptides, pharmaceutics, glycopolymers, glycosylated surfactants, or nucleic acids are all interesting systems that will lend themselves to similar investigations of their respective interactions with model membranes. There have been three major developments within the recent past: (i) GIXD measurements of monomolecular protein sheet crystals anchored at the air/water interface have become feasible45 and have been further developed to become an ever more powerful tool of surface characterization.12,46 (ii) X-Ray reflectivity measurements are boosted by the increased brilliance of modern synchrotron radiation sources and—to a lesser extent—of technical improvements of laboratory scale X-ray sources;12,22 at the same time, contrast variation with neutron reflection experiments and composition-refinement techniques for data evaluation,23,35,47 as well as model-free data inversion techniques24 and high-resolution models of lipid monolayers48 are taking advantage of such performance improvements. (iii) Finally, the FT-IRRAS technique for the investigation of monomolecular surface layers on aqueous subphases has taken the step beyond methylene spectroscopy of alkyl or acyl chains at the interface and has matured into a stable and ultra-sensitive technique for probing the molecular environment of specific molecular fragments within molecular surface architectures, yielding new insights both in life science and materials science.32,49–51 We have shown in this paper that the combination of these powerful methods enable the discussion of the structure of organic interface layers in molecular detail.Acknowledgements
We thank Jan Skov Pedersen (J.S.P.) for making available to us his program for form-free fitting of reflectivities24 and J.S.P. and Michael Gerstenberg for helpful advice. We are grateful to HASYLAB at DESY, Hamburg, for beamtime at the intense beamline BW121 of the DORIS bypass under contract no. II-97-51. This work has been supported by the German Science Foundation (SFB 294, TP C10), the Austrian Science Foundation (projects S7204/S7205), the Fonds der Chemischen Industrie, Frankfurt, the Danish Dansync programme, and the EC-TMR (contract no. ERBFMGECT950059).References
- A. DiederichM. Lösche, in Protein array: An alternate biomolecular system, ed. K. Nagayama, Japan Scientific Societies Press/Elsevier, Tokyo/Limerick, 1997, p. 205. Search PubMed.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117 CrossRef CAS.
- W. KnollM. LileyD. PiscevicJ. SpinkeM. J. Tarlov, in Protein array: An alternate biomolecular system, ed. K. Nagayama, Japan Scientific Societies Press/Elsevier, Tokyo/Limerick, 1997, p. 231. Search PubMed.
- M. Lösche, Curr. Opin. Solid State Mater. Sci., 1997, 2, 546. Search PubMed.
- E. Sackmann, Science, 1996, 271, 43 CrossRef CAS.
- U. B. Sleytr, P. Messner, D. Pum and M. Sára, Angew. Chem., Int. Ed., 1999, 38, 1034 CrossRef CAS.
- Crystalline Bacterial Cell Surface Proteins, ed. U. B. Sleytr, P. Messner, D. Pum and M. Sára, Academic Press, San Diego/Austin, 1996. Search PubMed.
- U. B. Sleytr and T. J. Beveridge, Trends Microbiol., 1999, 7, 253 CrossRef CAS.
- D. Pum, M. Sára and U. B. Sleytr, J. Bacteriol., 1989, 171, 5296 CAS.
- P. Messner and U. B. Sleytr, Adv. Microbiol. Physiol., 1992, 33, 213 Search PubMed.
- M. Sára and U. B. Sleytr, Prog. Biophys. Mol. Biol., 1996, 65, 83 CrossRef CAS.
- M. Weygand, B. Wetzer, D. Pum, U. B. Sleytr, K. Kjaer, P. B. Howes and M. Lösche, Biophys. J., 1999, 76, 458 Search PubMed.
- D. Pum and U. B. Sleytr, Trends Biotechnol., 1999, 17, 8 CrossRef CAS.
- D. Pum, M. Weinhandl, C. Hödl and U. B. Sleytr, J. Bacteriol., 1993, 175, 2762 CAS.
- D. Pum and U. B. Sleytr, Thin Solid Films, 1994, 244, 882 CrossRef CAS.
- B. Wetzer, A. Pfandler, E. Györvary, D. Pum, M. Lösche and U. B. Sleytr, Langmuir, 1998, 14, 6899 CrossRef CAS.
- B. Schuster, D. Pum and U. B. Sleytr, Biochim. Biophys. Acta, 1998, 1369, 51 CrossRef CAS.
- B. Schuster, D. Pum, O. Braha, H. Bayley and U. B. Sleytr, Biochim. Biophys. Acta, 1998, 1370, 280 CrossRef CAS.
- U. B. Sleytr, M. Sára, S. Küpcü and P. Messner, Arch. Microbiol., 1986, 146, 19 CrossRef CAS.
- A. Diederich, C. Sponer, D. Pum, U. B. Sleytr and M. Lösche, Colloids Surf. B, 1996, 6, 335 CrossRef CAS.
- R. Frahm, J. Weigelt, G. Meyer and G. Materlik, Rev. Sci. Instrum., 1995, 66, 1677 CrossRef CAS.
- P. KrügerM. SchalkeT. GutberletM. Lösche, in preparation..
- M. Lösche, M. Piepenstock, A. Diederich, T. Grünewald, K. Kjaer and D. Vaknin, Biophys. J., 1993, 65, 2160 Search PubMed.
- J. Skov Pedersen and I. W. Hamley, Physica B, 1994, 198, 16 CrossRef.
- I. W. Hamley and J. Skov Pedersen, J. Appl. Crystallogr., 1994, 27, 29 CrossRef CAS.
- J. Skov Pedersen and I. W. Hamley, J. Appl. Crystallogr., 1994, 27, 36 CrossRef CAS.
- J. Als-NielsenK. Kjaer, in Phase Transitions in Soft Condensed Matter, ed. T. Riste and D. Sherrington, Plenum Press, New York, 1989, p. 113. Search PubMed.
- J. Als-Nielsen, D. Jacquemain, K. Kjaer, M. Lahav, F. Leveiller and L. Leiserowitz, Phys. Rep., 1994, 246, 251 CrossRef CAS.
- G. H. Vineyard, Phys. Rev. B, 1982, 26, 4146 CrossRef CAS.
- K. Kjaer, Physica B, 1994, 198, 100 CrossRef CAS.
- R. A. Dluhy and D. G. Cornell, J. Phys. Chem., 1985, 89, 3195 CrossRef CAS.
- R. Mendelsohn, J. W. Brauner and A. Gericke, Annu. Rev. Phys. Chem., 1995, 46, 305 CrossRef CAS.
- C. R. Flach, A. Gericke and R. Mendelsohn, J. Phys. Chem., 1997, 101, 58 CrossRef CAS.
- D. A. CadenheadF. Müller-LandauB. M. J. Kellner, in Ordering in Two Dimensions, ed. S. K. Sinha, Elsevier North Holland, Amsterdam, 1980, p. 73. Search PubMed.
- M. WeygandB. WetzerD. PumU. B. SleytrP. B. HowesK. KjaerM. Lösche, 1999, in preparation..
- D. Vaknin, J. Als-Nielsen, M. Piepenstock and M. Lösche, Biophys. J., 1991, 60, 1545 Search PubMed.
- U. P. FringeliH. H. Günthard, in Membrane Spectroscopy, ed. E. Grell, Springer, New York, 1981, p. 270. Search PubMed.
- R. A. Dluhy, D. G. Cameron, H. H. Mantsch and R. Mendelsohn, Biochemistry, 1983, 22, 6318 CrossRef CAS.
- C. R. Flach, J. W. Brauner and R. Mendelsohn, Biophys. J., 1993, 65, 1994 Search PubMed.
- H. Hauser, I. Pascher, R. H. Pearson and S. Sundell, Biochim. Biophys. Acta, 1981, 650, 21 CAS.
- G. CevcD. Marsh, Phospholipid Bilayers, Physical Principles and Models, Wiley-Interscience, New York, 1987. Search PubMed.
- M. Sára, personal communication..
- S. J. Perkins, in Modern Physical Methods in Biochemistry, Part B, ed. A. Neuberger and L. L. M. V. Deenen, Elsevier, Amsterdam, 1988, p. 143. Search PubMed.
- M. Lösche, Habilitationsschrift [in German], Mainz University, 1994..
- H. Haas, G. Brezesinski and H. Möhwald, Biophys. J., 1995, 68, 312 Search PubMed.
- S. A. W. Verclas, P. B. Howes, K. Kjaer, A. Wurlitzer, M. Weygand, G. Büldt, N. A. Dencher and M. Lösche, J. Mol. Biol., 1999, 287, 837 CrossRef CAS.
- D. Vaknin, K. Kjaer, J. Als-Nielsen and M. Lösche, Biophys. J., 1991, 59, 1325 Search PubMed.
- M. SchalkeP. KrügerM. WeygandM. Lösche, Biochem. Biophys. Acta., 1999, in press. Search PubMed.
- A. Gericke, C. R. Flach and R. Mendelsohn, Biophys. J., 1997, 73, 492 Search PubMed.
- C. R. Flach, A. Gericke, K. M. W. Keough and R. Mendelsohn, Biochim. Biophys. Acta, 1999, 1416, 11 CrossRef CAS.
- M. SchalkeJ. FriedmannU. B. SleytrM. Lösche 1999, in preparation..
Footnotes |
| † Basis of a presentation given at Materials Chemistry Discussion No. 2, 13–15 September 1999, University of Nottingham, UK. |
| ‡ Current address: University of Leicester, Department of Physics and Astronomy, Leicester, UK LE1 7RH. |
| § Current address: Rhône-Poulenc Rorer, F-94403 Vitry-sur-Seine, France. |
| ¶ Phospholipid phases are denoted according to Cadenhead et al.34 |
| || pI = 4.3 is the isoelectric point of the S-layer protein from B. coagulans E38-66/V1. |
| This journal is © The Royal Society of Chemistry 2000 |