Tunable plasma sources in analytical spectroscopy: Current status and projections
R. Kenneth Marcusa, E. Hywel Evansb and Joseph A. Carusoc
aDepartment of Chemistry, Clemson University, Clemson, SC 29634-1905, USA
bDepartment of Environmental Sciences, University of Plymouth, Drake Circus, Plymouth, UK PL4 8AA
cDepartment of Chemistry, University of Cincinnati, Cincinnati, OH 45221-0721, USA
First published on UnassignedUnassigned7th January 2000
Abstract
While the developments in spectrochemical instrumentation have (for the most part) kept pace with the quantitative challenges of a diversity of applications, the qualitative aspects of spectrochemical analysis lag far behind. This is, of course, the principal challenge of ‘chemical speciation'. It seems clear at this point that a paradigm shift is in order if the atomic spectrometry community is to step up and solve the practical problems presented by biochemists, nutritionists, geologists, and environmental chemists. We present here an outline for progress in this area along with a summary of recent developments in the area of tunable plasma sources.
For the last four decades, the term plasma spectrochemistry has been synonymous with atomic spectrometry and methods for performing elemental analysis. Plasma sources such as the dc arc, high voltage spark, inductively coupled plasma (ICP), microwave-induced plasma (MIP), and glow discharge (GD) have each established their own niches with regard to specific analytical applications. By the same token, there are still a large number of elemental analyses performed by flame and electrothermal (furnace) atomic absorption spectrometry. Over the last 15 years or so, a growing amount of research effort has been focused on the development of methods of sample introduction for spectrochemical analysis, with actual source developments taking a more minor role in the literature and at scientific meetings. In fact, the focus of sample introduction has evolved from methods of enhancing sample utilization (i.e., new nebulizer systems) to coupling chromatographic separations to plasma sources. The driving force for this progression is simple to identify: chemical problems in many fields can be less dependent on elemental identity than on the chemical form of that element. Thus, these situations dictate that elemental identity and concentration information must be augmented with the identity of the chemical species. For complex (real world) samples, species-specific chemistries can at least allow the assignment of a number of compounds containing the element of interest. The use of chemical separation methods prior to element-specific detection is generically termined ‘elemental speciation'.
The coupling of liquid chromatography with inductively coupled plasma atomic emission and mass spectrometries (ICP-AES/MS) presents a powerful tool for the determination of elemental species present in a given eluting fraction. Particularly with mass spectrometric detection, very low levels of detectability can be realized and thus many important problems can be solved. Perusal of the topics covered in the elemental speciation literature illustrates potential impact in areas including nutrition, toxicology, geochemistry, and environmental chemistry. Fig. 1 illustrates the general concept of an elemental speciation experiment as is typically practiced with an element-specific detector. On a first principles basis, some form of chromatography is employed to separate components of a mixture based on interaction between the analyte and a stationary phase, leading to species-specific retention on the chromatographic column. The most common types of interactions are based on solvent–solute polarity in liquid chromatography (LC), electrostatic interactions in ion chromatography (IC), ion mobility in capillary electrophoresis (CE), and for gas chromatography (GC) solute volatility coupled with stationary phase affinity. Upon separation, analyte species are transported in the mobile phase through the sample introduction port of the spectrochemical device (source). With GC, the total column flow is often directed to the source, wherein mixing with the gas that sustains the source occurs and the analyte is subjected to the collisional/thermal processes in the source. For liquid phase separations, a phase change must occur to get the analyte species into the vapor phase. As such, some form of nebulizer/spray chamber combination is employed to generate (and possibly desolvate) an aerosol. Alternatively, direct injection nebulizers (DIN), wherein the entire desolvation/vaporization process occurs in the plasma, have found growing use with ICP sources. The advantage here is for higher efficiency utilization of minute samples under very low flow conditions, though this nebulizer is not compatible with normal LC flow rates.
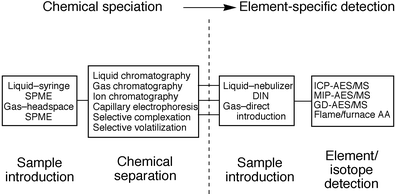 | ||
| Fig. 1 Basic elements of an elemental speciation experiment. | ||
Once the coupling of GC, LC/IC, or CE separations with traditional spectrochemical sources is achieved, the experiment becomes a simple time-resolved elemental analysis. That is, the spectrometer output for a given analyte element is plotted as a function of elution time. The method of analysis (AA, AES, or MS) and the precise configuration of the instrument available dictate the type of data obtainable and the ability to monitor single or multiple elements in real time. With mass spectrometry, a higher level of information may be obtained by monitoring specific isotopes of a given element. The result of a given experiment is an element/isotope-specific chromatogram. Herein lies the shortcoming of the vast majority of elemental speciation experiments; there is no direct information as to the chemical form of the analyte element, only an indication of which fractions contain atoms of the elements of interest. The identity of a given compound can only be derived via reference to retention times obtained in well characterized and matched separations of target species. This is of course a very tenuous situation in the case where retention time standards are not available or where variations in chromatographic conditions are present.
Fig. 2 illustrates a scale of information that might be desirable in, for example, the speciation analysis of an environmental sample. Beyond basic elemental analysis, the determination of a hydrated metal's oxidation state can provide many clues regarding environmental transport and, in some instances, relevant toxicological markers. Based on simple electrostatic interactions, the assignment of oxidation state (e.g., Fe2+vs. Fe3+) is easily done through ion chromatography retention times. However, this is only true for purely inorganic (aqueous) systems. The presence of additional ligated metals makes the assignment of precise identity and chemical consequence impossible without greater levels of information beyond the identity of the central metal atom. In these applications, the ability to identify the ligand species associated with each metal-containing eluent is very useful. For example, assignment of ligand class such as alkyl or aromatic organic functional groups can be an excellent clue to a compound's identity. Barring direct spectroscopic identification of a specific ligand, the ability to perform an empirical formula determination is useful. As a simplistic example, concentration ratios of carbon-to-hydrogen (C∶H) over the range from 1∶4 to 1∶1 indicate increasing levels of conjugation (multiple bonding); i.e., apliphatic to aromatic. As will be described in the following paragraph, this is the level of speciation information currently (commercially) available from the Hewlett-Packard GC-AED system. The ultimate elemental speciation information would allow exact identification of a chromatographic eluent, including central metal ion, ligand groups, and molecular formula. Thus, the goal of analytical spectroscopists should be the development of a spectrochemical device which is directly compatible with a given mode of chemical separation along with all of its variabilities (e.g., solvent make-up in LC), providing comprehensive chemical species information. A plasma-based instrument that achieves these goals does not exist—yet.
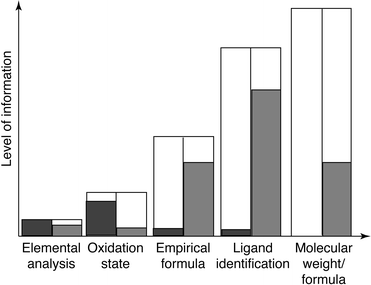 | ||
Fig. 2 Level of information desired in elemental speciation experiments. Filled regions represent relative amount of information currently available from commercial plasma source instrumentation for liquid ( ) and gaseous ( ) and gaseous ( ) sample introduction. ) sample introduction. | ||
Consideration of how the desired level of molecular information can affect the choice of a chromatographic detector is easily demonstrated for gas chromatography. The most straightforward detector is the flame ionization detector; it is also the most generic. Combustion of hydrocarbons eluting from the column produces a burst of electrons representative of the concentration of the compound, but does not provide any direct chemical information beyond what can be extrapolated based on retention times of standards. A higher level of information can be obtained from the electron capture detector. Here, radioactive decay of 63Ni produces a flux of low energy electrons (β-particles) which ionize N2 in the carrier gas resulting in a large population of electrons. Such low energy electrons are effectively ‘captured' by elements of high electronegativity (e.g., S, N, O, and halogens). In this case, detector signals are only observed for compounds containing atoms of those elements. As such, the detector is blind to simple hydrocarbons. Electron capture is sometimes referred to as a class-specific detector. Element-specific detection in gas chromatography can be obtained from the atomic emission of decomposed compounds in a low power helium MIP. This is the premise of the Hewlett-Packard GC-AED. In this case, the elemental composition and empirical formula of an eluting compound can in principle be derived through the relative emission intensities of the constituent atoms. Because the plasma environment is free from atmospheric gases, elements such as N, S, and O are viable analytes. Based simply on consideration of information content, the GC-AED is the most powerful of the plasma-based speciation tools. The greatest amount of chemical information obtainable in GC detection is via mass spectrometry, specifically electron impact ionization (EI). EI mass spectra of volatile organics and organometallics are generally classified as being moderate-to-hard in character. For some compounds, the spectra are dominated by molecular ions, while the spectra of more fragile compounds can show appreciable amounts of fragmentation. In some cases, very little of the ion species representing the molecular weight of the analyte are observed. In most instances, there is a molecular ion signature for each eluting compound, with sufficient fragmentation to assign a structure to the molecule. This is the underlying basis for the incredible diversity and power of GC-MS. In comparison with any sort of plasma source detectors, the very low cost and ease of operation of EI-GC-MS are also major factors in the success of the technique.
If all samples which required elemental speciation had sufficient volatility to perform GC separations, then GC-AED and GC-MS could solve all of the problems at hand. This is of course not the case. First, the vast majority of ‘speciation' samples are non-volatile and are in more or less aqueous media. Second, many speciation analytes are themselves non-volatile and thus would require some form of derivatization to be amenable to GC. Third, the chemical means of performing the separation (e.g., electrostatic interactions in IC) do not exist for gaseous specimens. Thus, it is clear that new paradigms (i.e., plasma sources) must be developed to accomplish comprehensive elemental speciation for liquid-phase separations. In reality, a middle ground in detector characteristics is also required for GC analyses. That is, there is a need for sources which provide a combination of the capabilities of GC-AED and GC-MS with electron ionization. In terms of challenges that are actually present in the wide array of elemental speciation applications, present methods are lacking relative to information content, ease of use, and capital costs. In reality, the vast majority of practitioners of elemental speciation tend to be atomic spectroscopists who see speciation as a subset of sample introduction, rather than chromatographers, biochemists, and environmental chemists who need this information.
The concept of tunable plasma sources comes about from the need to provide chemical information ranging from the elemental to the molecular level. The means by which spectrochemical plasma (both ICPs for liquids and GDs for solids) operate in an elemental analysis mode are by nature intended to destroy totally the molecular structure of analyte compounds. This is a requirement if the highest elemental sensitivity is to be achieved. Total atomization is also desirable from a spectroscopic point of view as molecular species can pose appreciable interferences in both optical and mass spectra. In elemental analysis, and purely inorganic speciation (e.g., Fe2+vs. Fe+3), the present approaches employing ICP-AES/MS or atomic absorption with very high temperature sources operating in reactive (atmospheric pressure) environments are adequate. To achieve molecular level speciation information, lower temperatures and inert atmospheres must be employed. While the reasoning for the former requirement is rather straightforward, the latter is more subtle The problems are two-fold. First, if one wishes to obtain information dealing with the ‘gaseous', non-metal elements in a compound (such as provided with the GC-AED), those species cannot be present in the plasma gas, the environment, or from other sources. For example, residual water vapor in a spectrochemical source yields a background of oxygen and hydrogen. Entrained air is equally detrimental. Second, for mass spectrometry, residual vapors of any sort will lead to deleterious ion–molecule reactions, greatly complicating the spectra and making interpretation, even of molecular ion identity, very difficult.
If the atomic spectrometry community is to meet the real needs of chromatographers, biochemists, environmental chemists, etc., new spectrochemical sources must be developed. It is generally true that a tunable plasma source used for elemental speciation will operate at lower powers (kinetic temperatures) than their elemental analysis counterparts. In addition, the source must operate in an inert environment, though not necessarily at reduced pressure. Only in these situations can the desired information (i.e., central metal, ligand species, empirical formula, and molecular formula) be obtained. Such a source could be developed as a variation of a presently employed device or perhaps be created on a first-principles basis. It must be kept in mind that the development of detectors which provide the desired information is not the only requirement of an integrated speciation instrument. Other practical concerns include:
Compatibility with the separation (mobile phase)
Ability to operate over normal ranges in mobile phase flow rates and compositions (e.g., buffers in LC)
Freedom from matrix effects as a function of mobile phase composition
Low cost
Ease of operation
Throughout any sort of development, the spectroscopist must be cognizant of the fact that persons presently involved in fields wherein speciation is needed are looking for chromatographic detectors to solve real problems. As such, the elemental speciation device must be designed more as an appendage to a chromatograph, rather than a chromatograph used as a means of sample introduction.
The purpose for organizing this special issue of the Journal of Analytical Atomic Spectrometry is to highlight current developments with non-atmospheric pressure Ar ICPs with an emphasis on tunable plasma sources. These could in principle be employed in elemental speciation applications. The following paragraphs review some of the significant work to date and thus set the stage for the individual contributions. It is hoped that this collection of work can serve as a benchmark and stimulus to atomic spectroscopists to use their backgrounds to develop more information-rich detectors for the very important applications outlined above.
Tunable plasma sources for atomic and molecular mass spectrometry
Inductively coupled plasmas (ICPs) have been used extensively as ion sources for elemental mass spectrometry (MS). Other plasmas such as microwave-induced plasmas (MIPs) and glow discharges (GDs) have been used less commonly. Conventionally, procedures such as electron ionization (EI), chemical ionization (CI), fast atom bombardment (FAB), thermospray and electrospray have been utilized as ion sources for molecular MS. However, the appropriate ion source will depend on the analyte to be studied, the degree of fragmentation which is desired, and the sample form. The conclusion that can be drawn from this is that, to gain the appropriate amount of chemical information, it is necessary to use the correct ion source. It would be advantageous if a single ion source could fulfil all of the roles cited above, i.e., for sensitive and element-selective quantitative analysis, and for qualitative analysis of unknowns.Early work on the use of MIPs as a possible ‘soft' ionization source for molecuar MS was undertaken by Heppner,1 who investigated an MIP formed with helium or hydrogen at between 30 and 150 W forward power, and at a pressure of between 10 and 200 Torr, in conjunction with GC sample introduction and EI mass spectrometry in a tandem approach. Compounds such as hexane and toluene were injected into an MIP using GC, though no actual chromatography was performed. The compounds were almost totally destroyed in the MIP, and broken down to constituent elements, which recombined to form simple polyatomic species such as CH, CH4, and CO in the MIP tail-flame. These compounds were then extracted into a mass spectrometer where they were ionized using EI and analyzed mass spectrometrically. Subsequently, Poussel et al.2 utilized an MIP formed with argon, xenon or krypton, and maintained at a power of between 25 and 50 W, at pressures between 2 × 10−2 and 6 × 10−2 mbar. Compounds such as cyclohexane and dodecane were introduced into the tail-flame of the MIP discharge where they were fragmented and ionized. These species were extracted into a mass spectrometer through a skimmer cone of 1.2 mm diameter, and yielded spectra similar to those obtained using EI. Similar work was performed by Olson et al.,3 who obtained spectra for chlorobenzene and toluene which were comparable to EI spectra. They also introduced compounds through the base of the MIP discharge and found that the molecular ions disappeared, with a consequent increase in the intensity of the monoatomic ions. The determination of trace organic compounds in air, by an atmospheric gas sampling GC-MS-MS operated in negative ion mode, was performed by McLuckey et al.4 The discharge was sustained using the sampled air and molecular fragments of the analytes were observed and used for the identification of explosives. A detection limit of 0.4 pg for trinitrotoluene (TNT) in air was achieved by desorption from glass.
Lauritsen et al.5 used a microporous poly(propylene) membrane for the introduction of aqueous samples into a mass spectrometer. A GD was chosen as a means of providing the ionizing chemical species because the filament source initially used suffered from reduced lifetimes due to chemical reaction with water. The GD cell was constructed by simply placing a high voltage on the electron target of a conventional electron impact source. One wall of this source was formed by the sampling membrane, with the organic analytes being transported preferentially across the membrane wall. The ions observed were quasi-molecular ions consisting of [MH]+ and [M(H2O)H]+. Direct liquid sample introduction into a GD plasma has been performed by Carazzato and Bertrand.6 Samples were dissolved in acetonitrile–water (70 + 30) and were introduced at a rate of 3.1 µl min−1, with [M + H]+ ions and fragment ions being observed in the spectrum. Detection limits for both caffeine and adenosine were in the 100 pg range.
Atmospheric pressure ionization (API) sources have also been developed utilizing MIP7,8 and GD9 sources. In the former work, the headspace vapor of perfluorotributylamine and tetramethyltin was introduced into an MIP and fragment ions were produced; the degree of fragmentation could be varied by changing the MIP power and carrier gas flow. A direct injection nebuliser (DIN) was also used to introduce a solution of tributylammonium hydroxide at a flow rate of 20–30 µl min−1. In the latter study, LC-API was interfaced with ion trap time-of-flight (TOF)-MS. When the discharge was operated at atmospheric pressure in He, and 0.1–0.6 mA, M+ and (M + H)+ ions were predominant, but when operated at approximately 2 Torr and 0.1–1.0 mA, a variable degree of fragmentation was effected. Detection limits of 2–3 fmol were achieved for pyridine.
A metastable beam source has been investigated by Faubert et al.10 for the selective fragmentation and ionization of a series of organic compounds. Analytes were introduced into a metastable atom beam which was extracted from a glow discharge. In this way, the analytes were ionized via Penning-type collisions. The degree of fragmentation of the analytes varied when the noble gas was changed, according to the metastable energy level. For example, little fragmentation was observed using krypton, yielding spectra not dissimilar to those obtained by CI, whereas helium caused extensive fragmentation, similar to EI source MS. In a similar vein, Kohler and Schlunegger11 used a low pressure Penning ionization source to achieve a tunable degree of fragmentation for a wide range of aliphatic, aromatic and halogenated compounds. Analyte vapor was used to form a plasma in the source, and by altering the current across the Penning electrodes, it was possible to alter the spectra of the analytes. At low discharge currents (10 µA), spectra similar to EI-MS were obtained, whereas at higher currents (120 µA), the spectra consisted of elemental species and small molecular fragments.
The aforementioned experiments were performed by introducing the pure compound or headspace vapor into the respective ion sources, so were not representative of analysis at concentrations found in most real samples. The first report of molecular fragmentation of ultratrace level analytes with discrete sample introduction was by Evans et al.,12 who coupled a low pressure inductively coupled plasma (LP-ICP) with gas chromatography (GC) and mass spectrometry (MS). At 6–50 W power and 1 mbar He pressure the source produced molecular ions and fragmentation spectra similar to an EI source. At 150 W power and 10 mbar Ar pressure, complete dissociation, and hence element-selective detection, was achieved for a variety of organohalogen and organometallic compounds.
Subsequently, the groups of Evans and Caruso published a joint paper on the use of the LC-ICP and radiofrequency glow discharge (rf-GD) as ion sources for molecular and atomic mass spectrometry using GC sample introduction.13 Olson et al.14 investigated an rf-GD for molecular fragmentation. The design was improved by Belkin et al.,15 who used a smaller discharge volume to minimize band-broadening. With this interface, five organotin compounds were successfully separated and detected. The mass spectrum observed for each species contained molecular fragment ions which were similar to those obtained from an EI source. A preliminary investigation into the effect of plasma operating conditions on the intensity of these fragment ions showed that plasma power and pressure had little effect on the fragment ion intensities. This interface was further refined by introducing the GC capillary co-axially to the cathode.16 This method of interfacing the GC capillary was seen to improve the signal stability by reducing the discharge asymmetry and preventing inhomogeneous sampling.
A novel method of sample introduction into low pressure GDs was reported by Gorecki et al.17 The sample of tetraethyllead was first extracted and adsorbed onto a solid-phase microextraction cartridge by sampling the headspace of a spiked water sample. The rf-GDMS source used for this study was similar to that reported by Belkin et al.15 After extraction, the solid-phase microextraction fiber was injected into a small oven wherein the volatile analyte was flash-vaporized. The desorbed tetraethyllead was carried to the GD source using the plasma gas. Detection of the analyte was achieved by observing the lead atomic ion signals and molecular fragment ion signals of tetraethyllead. Tentative detection limits were reported to be ca. 0.04 pg ml−1 as lead in water.
Despite the obvious potential of plasma sources for atomic and molecular mass spectrometry, one of the problems encountered by all workers is the tendency for molecular fragment ions to form only when a large mass of analyte is injected,12–17 typically between 5 and 50 ng. In order to compete with established techniques such as EI or CI, gaseous detection limits of between 0.005 and 0.05 ng are required. O'Connor et al.18 addressed this problem by introducing a reagent gas into the plasma. In this study it was observed that a helium plasma produced only elemental ion signals; however, the introduction of isobutane enabled the plasma to be operated in a tunable mode. The addition of 0.07 ml min−1 of isobutane to a 3 ml min−1 helium plasma produced mass spectra similar to EI spectra, for a series of organohalide compounds. On the addition of 0.2 ml min−1 of isobutane to the LP-ICP, only the molecular ion of the analyte compound was visible with detection limits between 2 and 200 pg µl−1 for the molecular ions. They defined the two modes of operation as ‘atomic' (i.e., element-selective detection) and ‘molecular' (i.e., qualitative analysis of unknowns). The same authors optimized the system for the determination of tetraethyllead in fuel,19 and achieved detection limits of 7 pg µl−1 for the total ion signal, and 20 and 70 pg µl−1 for the fragment ions at m/z 208 and 295, respectively, when operating in the ‘molecular' mode. It was possible to match almost exactly the mass spectrum for tetraethyllead in NIST SRM 1637 II Gasoline with an EI source library spectrum. Recently, Guzowski et al.20 have utilized a GD source for atomic and molecular mass spectrometry with gaseous sample introduction via an exponential dilutor. They achieved detection limits of between 24 and 93 pg s−1 for a range of halogenated organic compounds in atomic mode, and molecular fragmentation mass spectra when a reagent gas was added, though no detection limits were given in this mode of operation.
References
- R. A. Heppner, Anal. Chem., 1983, 55, 2170 CrossRef CAS.
- E. Poussel, J. M. Mermet, D. Deruaz and C. Beaugrand, Anal. Chem., 1988, 60, 923 CrossRef CAS.
- L. K. Olson, C. W. Story, J. T. Creed, W. Shen and J. A. Caruso, J. Anal. At. Spectrom., 1990, 5, 471 RSC.
- S. A. McLuckey, G. L. Glish, K. G. Asano and B. C. Grant, Anal. Chem., 1988, 60, 2220 CrossRef CAS.
- F. R. Lauritsen, T. K. Choudury, L. E. Dejarme and R. G. Cooks, Anal. Chim. Acta., 1992, 266, 1 CrossRef CAS.
- D. Carazzato and M. J. Bertrand, J. Am. Soc. Mass Spectrom., 1994, 5, 305 CrossRef CAS.
- W. Shen and R. D. Satzger, Anal. Chem., 1991, 63, 1960 CrossRef CAS.
- N. P. Vela, J. A. Caruso and R. D. Satzger, Appl. Spectrosc., 1997, 51, 1500 Search PubMed.
- B. M. Chien, S. M. Michael and D. M. Lubman, Anal. Chem., 1993, 65, 1916 CrossRef CAS.
- D. Faubert, G. J. C. Paul, J. Giroux and M. J. Bertrand, J. Mass Spectrom. Ion Processes, 1993, 124, 69 Search PubMed.
- M. Kohler and U. P. Schlunegger, J. Mass Spectrom., 1995, 30, 134 CAS.
- E. H. Evans, W. Pretorius, L. Ebdon and S. Rowland, Anal. Chem., 1994, 66, 3400 CrossRef CAS.
- G. O'Connor, L. Ebdon, E. H. Evans, H. Ding, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 1151 RSC.
- L. K. Olson, M. Belkin and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 491 RSC.
- M. Belkin, L. K. Olson and J. A. Caruso, J. Anal. At. Spectrom., 1997, 12, 1255 RSC.
- M. Belkin, J. W. Waggoner and J. A. Caruso, Anal. Commun., 1998, 35, 281 RSC.
- T. Gorecki, M. Belkin, J. A. Caruso and J. Pawlyszyn, Anal. Commun., 1997, 34, 275 RSC.
- G. O'Connor, L. Ebdon and E. H. Evans, J. Anal. At. Spectrom., 1997, 12, 1263 RSC.
- G. O'Connor, L. Ebdon and E. H. Evans, J. Anal. At. Spectrom., 1999, 14, 1303 RSC.
- J. P. Guzowski, J. A. C. Broekaert, S. J. Ray and G. M. Hieftje, J. Anal. At. Spectrom., 1999, 14, 1121 RSC.
| This journal is © The Royal Society of Chemistry 2000 |