A solvent-free Jacobs–Gould reaction
Teresa Cablewski, Paul A. Gurr, Peter J. Pajalic and Christopher R. Strauss*
CSIRO Molecular Science, Bag 10, Clayton South, Victoria 3169, Australia
First published on UnassignedUnassigned8th February 2000
Green ContextThere are a limited number of suitable solvents for thermal processes such as the Jacobs–Gould reaction which can be used to form the quinolone ring system of quinolone antibacterials. High temperature solvents such as biphenyl and diphenyl ether are solids under ambient conditions, which complicates work-up and recovery. Ideally such reactions would be carried out in the absence of solvent which would simplify the procedure and minimise waste. In this article an effective solvent-free thermal reaction is described which leads to multiple green chemistry improvements, notably greater energy efficiency, simpler work-up and waste minimisation. The process has also been successfully extended to a continuous system.JHC |
Summary
The Jacobs–Gould intramolecular cyclisation of diethyl N-(6-methyl-2-pyridyl)aminomethylenemalonate (1) to 3-ethoxycarbonyl-7-methyl-1,8-naphthyrid-4-one (2) was conducted without solvent. Empirical kinetic data were extrapolated to determine optimal reaction conditions. In contrast with established methods employing heat transfer oils, the reaction proceeded in high conversion, rapidly, predictably and controllably, at temperatures near 400 °C. Since dilution was unnecessary, the procedure facilitated high throughput, was energy efficient, low polluting and offered convenient work-up. A continuous Jacobs–Gould reaction (of 1 to 2) was demonstrated for the first time.Introduction
Oils derived from biphenyl and diphenyl ether are commonly employed as diluents and heat transfer media for reactions at temperatures above 180 °C, and typically around 250 °C.1 However, solidification of the individual components (biphenyl, mp 69–71 °C; diphenyl ether, mp 26–30 °C) or the eutectic mixture (mp 12 °C), can complicate isolation of products, particularly on the industrial scale. Losses of oil occur during recovery and purification, owing to penetration through valves, pumps and flanges and by evaporation through vents.2 The vapours are disagreeable even at concentrations below 10 ppm and acceptable replacement oils are sought.3However, a limited range of alternatives has led to such oils being used extensively for thermal processes e.g. in Jacobs–Gould reactions,4 by which the quinolone ring systems of quinolone antibacterials can be formed.1,5,6 To explore the feasibility of avoiding heat transfer oils, we have investigated the cyclisation of diethyl N-(6-methyl-2-pyridyl)aminomethylenemalonate (1) to 3-ethoxycarbonyl-7-methyl-1,8-naphthyrid-4-one (2), which is the key intermediate toward the first commercial quinolone antibacterial, nalidixic acid (3; see Scheme 1).5
 | ||
| Scheme 1 | ||
Results and discussion
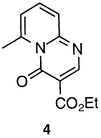
The roles of solvents, their function and selection criteria for environmentally benign chemistry were recently reviewed by Nelson.7 For intramolecular transformations in particular, high dilution is employed to inhibit competing intermolecular processes. Clearly, if achievable, solvent-free intramolecular processes would be preferable.To explore that possibility, neat methylenemalonate 1 was heated in Pyrex glass vessels for up to 50 min at 260, 270, 280, 290 and 300 °C. Low heating rates and temperatures facilitated formation of significant proportions of the pyrimidinone by-product 4 by an alternative intramolecular ring closure. The highest conversion to naphthyridone 2 was 44% at 300 °C, after 5 min.8 Although the maximum conversion increased with reaction temperature, extended heating, even at 260 °C, led to decomposition.
From these experiments, the times required to achieve 10%, 20%, 30% and 40% conversions were plotted as a function of temperature (see Fig. 1; semi-log scale). Extrapolation of the reaction times indicated the shorter periods and higher temperatures required for comparable conversions. These conditions were verified experimentally, as shown by data points for temperatures above 300 °C. Also, the plots for 30 and 40% conversion were more nearly co-linear than the 10 and 20% plots in Fig. 1. This suggested that as the yield increased, relatively minor changes in temperature and time substantially affected the outcome. A conversion of 40% or greater was only obtained at 290 °C and above (note that the 30% and 40% conversion plots do not meet the y-axis in Fig. 1) and decomposition occurred with longer heating times, so the useful heating period and temperature range both narrowed with increasing yields. These results indicated that optimal conditions would lie near the bottom right hand corner of Fig. 1, with temperatures of at least 370 °C and reaction times of less than 1 min.
 | ||
| Fig. 1 Plots of reaction time against temperature for 10, 20, 30 and 40% conversions of 1 to 2. | ||
When such conditions were employed, greater conversions (over 80%) to 2 were obtained than over any time interval explored at lower temperatures. High precision was necessary, as the product polymerised if the heating was continued for even seconds beyond the required time. The optimal conditions (45 s at 385 °C) afforded 2 in 86% conversion.
The short heating periods enabled adaptation of the process for flowthrough operation. To our knowledge, this is the first example of a continuous Jacobs–Gould reaction and 2 was obtained in 79% conversion (see experimental).
From this work, Jacobs–Gould reactions can proceed in high conversion, rapidly, predictably and controllably, without a diluting heat transfer medium. Surprisingly, the optimal conditions involved considerably greater temperatures than have been employed traditionally. Since dilution was unnecessary, far less processing than before was needed to obtain a similar amount of product. Hence, the procedure was energy efficient, low polluting, convenient to work-up and afforded high throughputs of reactant. Interestingly, the mixture of biphenyl and diphenyl ether slowly decomposes near 400 °C and so it could not be used routinely at the optimal reaction temperatures anyway.2
Naphthyridone 2 has a high melting point and is insoluble in common solvents. Such characteristics are typical for crystalline compounds with extensive hydrogen bonding. Since recrystallisation was impractical, crude 2 was triturated and the impurities were leached at room temperature by acetone, diethyl ether or ethyl acetate, all of which could be recycled. By this process, 2 could be obtained in high purity.
Quinolone antibacterials constitute an important commercial class of antibiotics and there are now several such compounds on the market, with others undergoing clinical trials.9 Some have a broader spectrum and are at least an order of magnitude more potent than nalidixic acid 3. We have applied the developed methodology to the preparation of 5 and 6. Both could serve as intermediates for analogues. Although the conversions were lower, the results were consistent with those for the cyclisation of 1 to 2.
We hope that these results and the methodology involving the extrapolation of empirical kinetic data to determine optimal reaction conditions, will encourage investigation of further thermal intramolecular processes under solvent free conditions.
Experimental
General
General procedures have been described previously.10 Methylenemalonate 1 was produced by a literature method.11 Physical data for products or synthesised reference compounds, agreed with literature values.
Continuous process
Methylenemalonate 1 at 170 °C, was pumped at a rate of 14 g per min through a stainless steel coil (14.8 mL, 3.8 m long, 2.8 m of which was immersed) heated in a fluidised sand bath at 380 °C. The product (effluent temperature 290 °C), which contained naphthyridone 2 (79%), 1 (8%), pyrimidinone 4 (5%) and unknown(s) (8%) was collected in chilled stainless steel vessels.A sample (20 g) of the crude product was ground with a mortar and pestle and suspended in Et2O (60 mL). The mixture was stirred at room temperature for 1.5 h and the solid removed by filtration. The leaching procedure was repeated twice more with Et2O (48 and 40 mL, respectively), to afford naphthyridone 2 (15.4 g) in 94% purity, by 1H NMR analysis.
References
- A. Pipaud, R. Rocher and J. Chenault, Synth. Commun., 1997, 27, 1927 CAS; K. Heleyova, D. Ilavsky, V. Bobosik and N. Pronayova, Collect. Czech. Chem. Commun., 1996, 61, 371 CrossRef CAS; K. Heleyova and D. Ilavsky, Collect. Czech. Chem. Commun., 1997, 62, 99 CrossRef CAS; T. O. Richardson, N. Neale and N. Carwell, J. Heterocycl. Chem., 1995, 32, 359 Search PubMed; M. Uchida, K. Otsubo, J. Matsubara, T. Ohtani, S. Morita and K. Yamasaki, Chem. Pharm. Bull., 1995, 43, 693 Search PubMed; D. Heber, U. Ravens and T. Schulze, Pharmazie, 1993, 48, 509 Search PubMed; J. C. Carretero, J. L. Garcia Ruano and M. Vicioso, Tetrahedron, 1992, 48, 7373 CrossRef CAS.
- W. J. Danziger, in Kirk-Othmer Encyclopedia of Chemical Technology, Second Edition, John Wiley, New York, 1966, vol. 10, p. 846. Search PubMed.
- J. Krieger, Chem. Eng. News, 1991, 69((51)), 17 Search PubMed.
- R. G. Gould and W. A. Jacobs, J. Am. Chem. Soc., 1939, 61, 2890 CrossRef CAS.
- R. Albrecht, Prog. Drug. Res., 1977, 21, 9 Search PubMed.
- S. Radl and D. Bouzard, Heterocycles, 1992, 34, 2143 Search PubMed.
- W. M. Nelson, in Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes, ed. P. T. Anastas and T. C. Williamson, Oxford University Press, Oxford, 1998, ch. 12, pp. 200–224. Search PubMed.
- In three experiments selected at random, conversions and isolated yields were comparable..
- D. Barrett, H. Sasaki, T. Kinoshita, A. Fujikawa and K. Sakane, Tetrahedron, 1996, 52, 8471 CrossRef CAS; I. Hermecz, L. Vasvari-Debreczy, B. Podanyi, G. Kereszturi, M. Balogh, A. Horvath and P. Varkonyi, Heterocycles, 1998, 48, 1111 and references cited therein.; Search PubMed; J. P. Sanchez, J. M. Domagala, C. L. Heifetz, S. R. Priebe, J. A. Sesnie and A. K. Trehan, J. Med. Chem., 1992, 35, 1764 Search PubMed; K. Grohe, Chem. Br., 1992, 28, 34 CrossRef CAS.
- T. Cablewski, P. A. Gurr, K. D. Raner and C. R. Strauss, J. Org. Chem., 1994, 59, 5814 CrossRef CAS.
- G. R. Lappin, J. Am. Chem. Soc., 1948, 70, 3348 CAS.
| This journal is © The Royal Society of Chemistry 2000 |