D-Gluconolactoneas a precursor to new environmentally benign tensioactive agents
Florence Garésioa, Nathalie Kardosa, Christian Bonneviea, Serge Petitb and Jean-Louis Luche*a
aLaboratoire de Chimie Moléculaire et
Environnement, Université de Savoie-ESIGEC, 73376, Le Bourget du Lac Cedex, France.. E-mail: Jean-Louis.Luche@univ-savoie.fr
bLaboratoire de Chimiométrie, Université Claude Bernard de Lyon, 43 Boulevard du 11 Novembre 1918, 69622, Villeurbanne Cedex, France.. E-mail: petit@cpe.fr
First published on UnassignedUnassigned8th February 2000
Green ContextSurfactants are used in great quantities in many applications, but suffer from several drawbacks. For example, their stability to biodegradation can often cause difficulties in the environment, and the reliance on fossil fuel as a source of raw materials is also a problem in the longer term. The development of novel surfactant structures derived from renewable sources is described in this article. The products are designed to degrade to materials without surfactant properties, making them less of a problem environmentally. Several experimental parameters are investigated with a view to optimising the environmental impact of the production of these new surfactants.DJM |
Summary
The conditions for the preparation of acetals with tensioactive properties from γ- and δ-gluconolactones and dodecanal and tetradecanal were studied. The role of the solvent, the catalyst and the influence of sonication on the yields (up to 60–80%) were examined, and a mechanistic interpretation is formulated.Introduction
The chemical stability of conventional tensioactive agents synthetised from fossil resources is at the origin of many environmental problems,1 making their replacement by eco-friendly substitutes highly desirable. This observation has prompted a research effort aimed at the exploitation of raw materials from renewable resources, and their transformation to new types of surfactants easily decomposed in the natural environment. A strategy was developed using a carbohydrate derivative as the polar hydrophilic part, and a fatty chain compound for the apolar part of the molecule.2 The link between the two parts can be, among others,3 an acid-labile acetal group (Scheme 1), and a number of compounds derived from such basic structures were shown to have excellent tensioactive properties. Cleavage to non-tensioactive fragments offers advantages both at the usage stage by permitting a progressive delivery of a bioactive substance, or the disposal due to their decomposition to products with minimised environmental impact. | ||
| Scheme 1 Acetalic carbohydrate-based tensioactive. | ||
Among the carbohydrates tested, sucrose itself can be ketalised with diverse carbonyl compounds.4 The reactivity is low, however, and indirect procedures from aldehyde dimethylacetals were used,5 in some cases under sonication,6 but even with this activation the yields remain modest. Another problem is the selectivity, due to the number of hydroxy groups, and protection steps are sometimes required.5 The acetals derived from the available and cheap glucono- 1 or galactonolactones, which can undergo a nucleophilic ring opening to introduce a second apolar chain,7 can be prepared in good yields,8,9 but some drawbacks persist such as long reaction times, high amounts of catalysts, and the requirement for expensive (DMF) or toxic (benzene) solvents. All these elements prompted us to undertake an optimisation of the reaction shown in Scheme 2. Previous work in this laboratory10 established that the experimental conditions published9 can be improved by replacing benzene by hexane. Nevertheless, a few other parameters (catalyst, solvent, time, ...) deserved examination to reach further improvements. This paper reports our recent findings.
 | ||
| Scheme 2 Acetalisation of D-gluconolactone 1. | ||
Results and discussion
First, we found that the toxic hexane can be changed for cyclohexane, and toluene-4-sulfonic acid for methanesulfonic acid (MsOH), which is known to exhibit interesting characteristics in terms of green chemistry.11 Thus, MsOH and cyclohexane have been systematically used in this work. Since the reaction medium is heterogeneous, all the experiments were duplicated under ultrasound irradiation.12It was found that the optimal solvent composition corresponds to a DMF∶cyclohexane ratio of 3∶17 (Table 1). A further decrease of the DMF amount (2∶18) slows down the process and degradations predominate. The sonochemical reactions provide satisfactory results, even if their advantage is not obvious. Despite extensive experimentation, reaction using octan-2-one gave the expected acetal in a disappointing 13–16% yield.
| Entry | Aldehyde | Solventa | Conditions | Yieldb (%) |
|---|---|---|---|---|
| a Solvent A = DMF–cyclohexane (5∶15, v∶v); Solvent B = DMF–cyclohexane (3∶17, v∶v).b Isolated yield. | ||||
| 1 | Dodecanal | A | Reflux, 4.5 h | 67 |
| 2 | Dodecanal | A | Reflux, 2.5 h | 69 |
| 3 | Dodecanal | A | Reflux, 0.5 h | 37 |
| 4 | Dodecanal | A | )))), 1.5 h | 79 |
| 5 | Dodecanal | A | )))), 0.5 h | 42 |
| 6 | Dodecanal | B | Reflux, 2 h | 79 |
| 7 | Dodecanal | B | Reflux, 0.5 h | 38 |
| 8 | Dodecanal | B | )))), 2 h | 74 |
| 9 | Dodecanal | B | )))), 0.5 h | 33 |
| 10 | Tetradecanal | B | Reflux, 2 h | 60 |
| 11 | Tetradecanal | B | Reflux, 0.5 h | 56 |
| 12 | Tetradecanal | B | )))), 2h | 61 |
| 13 | Tetradecanal | B | )))), 0.5 h | 47 |
A most important question is that of the acetal structure. In a previous study,10 we established that, contrary to literature data,9 the product is a dioxolane (1,4-DGLA, 2) derived from the 1,4-gluconolactone 3 and not the 1,5-lactone derivative (1,5-DGLA, 4) (Scheme 3).13
 | ||
| Scheme 3 Isomeric gluconolactone acetals 2 and 4. | ||
It can thus be envisaged that the formation of 2 occurs in two consecutive steps, a ring contraction followed by the acetalisation, making the lactone 3 a potentially better starting material. Therefore a second series of experiments was undertaken from this compound, prepared from the commercial lactone 1 by an acid catalysed ring contraction.14 This translactonisation (a transesterification) should most probably proceed by a AAC2 process via the bicyclic GAA 5 as the tetrahedral intermediate known to be involved in such mechanisms (Scheme 4).15
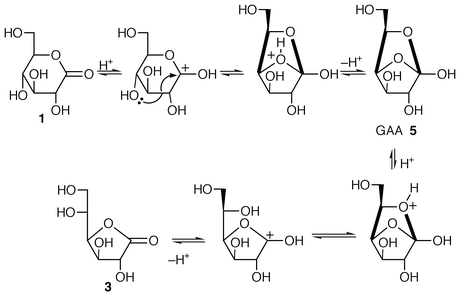 | ||
| Scheme 4 Mechanism proposal for the ring contraction of 1 to 3. | ||
Experiments run from 3 (Table 2) demonstrate that no obvious advantage results from its use to prepare 2 as compared to its isomer 1, in terms of rates and yields. These results should mean that the pathway does not necessarily involve the sequence ‘1 to 3 then 3 to 2‘, which led to the proposal of an alternative interpretation (Scheme 5).
| Entry | Aldehyde | Conditionsa | Yieldb (%) |
|---|---|---|---|
| a Solvent = DMF–cyclohexane (3∶17, v∶v) in all the cases.b Isolated yields | |||
| 1 | Dodecanal | Reflux, 2 h | 70 |
| 2 | Dodecanal | Reflux, 0.5 h | 65 |
| 3 | Dodecanal | )))), 2 h | 70 |
| 4 | Dodecanal | )))), 0.5 h | 38 |
| 5 | Tetradecanal | Reflux, 2 h | 60 |
| 6 | Tetradecanal | Reflux, 0.5 h | 47 |
| 7 | Tetradecanal | )))), 2 h | 45 |
| 8 | Tetradecanal | )))), 0.5 h | 34 |
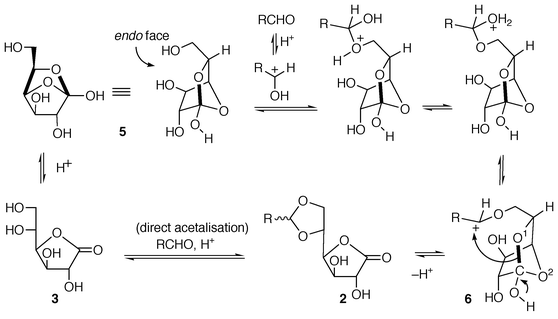 | ||
| Scheme 5 Mechanism proposal for the formation of 2. | ||
The key intermediate in these reactions should be 5, formally
an intramolecular acetal of gluconic acid, or a
‘hemi-orthoester’. Thus, the 1 to 3
isomerisation implies a simple opening of the 6-membered ring of
5, as shown in Scheme 4. On the
other hand, the reaction of a carbonyl compound with the latter compound is
actually a transacetalisation, the first step of which is the addition of
the primary alcohol to the protonated aldehydic C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O group. Further
steps consist in the elimination of water to give a carbocation which
rearranges to the acetal in a rather straightforward manner. Two
observations give substance to this proposal: (i) the steric crowding on
the endo face of the transient 5 explains the poor
reactivity of octan-2-one; (ii) the formation of 1,5-DGLA 4 is
disfavoured since it would require the cleavage of the C–O2 bond (see structure 6 in Scheme 5) to achieve the cyclisation on the
cationic site on the opposite side of the molecule. If the direct
acetalisation remains possible, it does not appear to be particularly
favoured in terms of rates in comparison to the other route.
O group. Further
steps consist in the elimination of water to give a carbocation which
rearranges to the acetal in a rather straightforward manner. Two
observations give substance to this proposal: (i) the steric crowding on
the endo face of the transient 5 explains the poor
reactivity of octan-2-one; (ii) the formation of 1,5-DGLA 4 is
disfavoured since it would require the cleavage of the C–O2 bond (see structure 6 in Scheme 5) to achieve the cyclisation on the
cationic site on the opposite side of the molecule. If the direct
acetalisation remains possible, it does not appear to be particularly
favoured in terms of rates in comparison to the other route.
To conclude the present study, the results to be underlined are: (i) the acetalisation of the two isomeric lactones 1 and 3 provides the same product 2, (ii) it is possible to achieve this transformation from commercially available materials, without the need to convert them to potentially more reactive compounds (including the use of aldehyde dimethylacetals), (iii) the use of a catalyst and solvents known to be more eco-friendly than in the previously published procedures, and (iv) the yields, up to ca. 80%. A more accurate view on the mechanism provides an unified explanation of the ring contraction leading to 3 and the acetalisation itself as well.
Experimental
D-Glucono-1,5-lactone 1 (from Aldrich, 1.07 g, 6 mmol, 1.2 equiv.), dodecanal (from Accros, 0.920 g, 5 mmol, 1 equiv.), MsOH (64.8 μL, 0.2 equiv.), and the solvents (total volume 20 mL) are refluxed (ca. 80 °C) in a round-bottom flask with a Dean-Stark water trap. The reaction is monitored by TLC. After completion and cooling, MsOH is neutralised with triethylamine (140 μL, 0.2 equiv.), the mixture is filtered through a 1∶1 silica–alumina pad (1 g each) and the solvents evaporated in vacuo. The residue is purified on a silicagel column, eluting with cyclohexane–ethyl acetate (2∶3) containing 1% NEt3.An alternative work-up can be used. After neutralisation, the two phases are separated. Water (10 mL) is added to the DMF (lower) layer, and the apolar products (residual aldehyde and other contaminants) are extracted with cyclohexane. The remaining milky suspension is then extracted with diethyl ether (3 × 15 mL). The ethereal phases are dried (MgSO4) and the solvent evaporated.
The yellowish solid obtained after one of these work-ups is recrystallised from cyclohexane, providing the 1∶1 mixture of stereoisomers of 2a as white crystals, mp 108–109 °C (lit. 105–107 °C);9a,16 ν(KBr)/cm−1 3455, 2923, 2849, 1754, 1470, 1245, 1220, 1215, 1100, 1065, 962. δH(200 MHz, DMSO-d6) 6.38 (dd, 1 H), 5.79 (dd, 1 H), 4.92 (t, 0.5 H), 4.80 (t, 0.5 H), 4.68 (t, 0.5 H), 4.57 (t, 0.5 H), 4.32 (m, 1 H), 4.2–3.69 (m, 4 H), 1.54 (m, 2 H), 1.25 (m, 18 H) and 0.86 (t, 3 H); δC(50.32 MHz, DMSO-d6) 175.2, 103.6, 103.5, 81, 73.5, 73.3, 72.7, 72.5, 72.1, 71.9, 65.9, 65.4, 33.3, 33.2, 31.2, 28.9, 28.7, 23.5, 23.4, 22.0 and 13.9. MS (electron impact) for C18H32O6: 344 (M+), 343, 227, 189, 160, 143 (100%). 2b from tetradecanal is obtained by the same procedures: mp 112–114 °C (lit., 112–115 °C);16 ν(KBr)/cm−1 3440, 2956, 2850, 1763, 1470, 1226, 1107, 1059 and 961. δH(200 MHz, DMSO-d6) 6.40 (dd, 1 H), 5.78 (dd, 1 H), 4.90 (t, 0.5 H), 4.80 (t, 0.5 H), 4.68 (t, 0.5 H), 4.55 (t, 0.5 H), 4.35 (m, 1 H), 4.25–3.70 (m, 4 H), 1.70–1.45 (m, 2 H), 1.45–1.10 (m, 22 H) and 0.85 (t, 3 H); δC(50.32 MHz, DMSO-d6) 175.2, 103.6, 103.5, 81.3, 73.5, 73.3, 72.7, 72.6, 72.1, 71.9, 65.9, 65.5, 33.3, 33.2, 31.3, 29.0, 28.9, 28.7, 23.4, 23.4, 22.0 and 13.9. MS (electron impact) for C20H36O6: 372 (M+), 371, 255, 189, 160, 143 (100%). The sonochemical experiments were performed in a glass vessel equipped with a condenser, using identical amounts of materials and solvents, with a Ultrason-Annemasse 30 kHz generator delivering an energy of 14 W, estimated calorimetrically.17
Acknowledgements
We thank IDE’ALP for financial support, and Professors G. Reverdy and C. Petrier for their interest in this work.References
- G. Jakobi and A. Löhr, Detergents and Textile Washing: Principles and Practice, VCH, Weinheim, 1987. Search PubMed.
- P. A. Egan, Chemtech., 1989, 758 Search PubMed.
- (a) J. Gagnaire, G. Toraman, G. Descotes, A. Bouchu and Y. Queneau, Tetrahedron Lett., 1999, 40, 2757 CrossRef CAS; (b) F. A. Hughes and B. W. Lew, J. Am. Oil Chem. Soc., 1970, 47, 162 Search PubMed; (c) Carbohydrates as Organic Raw Materials, ed. F. W. Lichtenthaler, VCH, Weinheim, 1991. Search PubMed.
- P. Salanski, G. Descotes, A. Bouchu and Y. Queneau, J. Carbohydr. Chem., 1998, 17, 129 Search PubMed.
- E. Fanton, C. Fayet and J. Gelas, Carbohydrate Res., 1997, 298, 85 CrossRef CAS.
- J. Besson, C. Fayet, J. Gelas and C. Lamazzi, OPPI Briefs, 1998, 30, 460 Search PubMed.
- C. L. Mehltretter, M. S. Furry, R. L. Mellies and J. C. Rankin, J. Am. Oil Chem. Soc., 1952, 202 Search PubMed.
- M. Csiba, J. Cleophax, A. Loupy, J. Malthète and S. D. Gero, Tetrahedron Lett., 1993, 34, 1787 CrossRef CAS.
- (a) T. Kida, A. Masuyama and M. Okahara, Tetrahedron Lett., 1990, 31, 5939 CrossRef CAS; (b) T. Kida, A. Morishima and Y. Nakatsuji, J. Am. Oil Chem. Soc., 1994, 71, 705 Search PubMed.
- (a) C. Bonnevie, PhD Thesis, Chambéry (France), 1998; (b) C. Bonnevie, D. Bouchu, F. Garésio, N. Kardos, G. Reverdy, P. Lanteri and S. Petit, Carbohydr Res., submitted. Search PubMed.
- M. D. Gernon, Min Wu, T. Buszta and P. Janney, Green Chem., 1999, 127 RSC.
- A. Loupy and J. L. Luche, Sonochemistry in Biphasic Systems, in Synthetic Organic Sonochemistry, ed. J. L. Luche, Plenum Press, New York, 1998, pp. 107–166. Search PubMed.
- Infrared, 1H (NOESY), 13C NMR, and mass spectra (presence of a peak at m/z [M−117]+, evidencing the cleavage of the C4–C5 bond) fit only with the 1,4-DGLA structure..
- (a) K. Bock, I. Lundt and C. Pedersen, Acta Chim. Scand. B, 1981, 35, 155 Search PubMed; (b) G. J. F. Chittenden, Recl. Trav. Chim. Pays Bas, 1988, 107, 455 Search PubMed.
- J. March, Advanced Organic Chemistry, Wiley, New York, 1992, 4th edn., p. 380. Search PubMed.
- S. Petit and S. Fouquay, Ger. Offen., DE 19,814,786 (Chem. Abstr., 1998, 129, 317972d)..
- T. J. Mason, J. P. Lorimer, D. M. Bates and Y. Zhao, Ultrason. Sonochem., 1994, 1, S91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |