Dynamic behaviour of 1,1,2-trichlorotrifluorethane on aluminium(III) chloride and related surfaces
David G. McBetha, Margaret M. McGeougha, Geoffrey Webb*a, John M. Winfield*a, Archie McCullochb and Neil Winterton†b
aDepartment of Chemistry, University of Glasgow, Glasgow, UK G12 8QQ. E-mail: J.Winfield@chem.gla.ac.uk
bResearch and Technology Department, ICI, The Heath, Runcorn, UK WA7 4QD
First published on UnassignedUnassigned8th February 2000
Green ContextThe safe destruction of CFCs is one of the major current issues in halocarbon chemistry. It seems likely that their production and consumption will be prohibited by the Montreal Protocol and related agreements. The remarkable volume of illegal traffic in these substances adds to the urgency to devise effective methods of destruction. Ideally their destruction should lead to other useful products and among the more likely are the more environmentally acceptable hydrofluorocarbons. This paper is concerned with one of the more important reactions that can be utilised in trying to achieve these goals—the isomerisation of 1,1,2-trichlorotrifluoroethane.JHC |
Summary
The isomerization of CCl2FCClF2 to CCl3CF3 is one of the steps in a possible scheme of reactions to convert CCl2FCClF2 to the more environmentally-acceptable refrigerant CF3CH2F. A reinvestigation of the isomerization in the presence of solid aluminium(III) chloride at ambient temperature indicates that the catalytically active sites for isomerization are generated in situ by the behaviour of aluminium(III) chloride as a chlorinating reagent towards CCl2FCClF2. Aluminium(III) chloride pretreated with CH3CCl3 exhibits very similar behaviour but γ-alumina chlorinated with CCl4 shows little or no isomerization activity.Introduction
One of the imperatives that underlie current research in the halocarbon field is the need to devise suitable methods for the destruction or conversion of compounds such as chloro- and bromo-fluorocarbons (CFCs and Halons) whose production and consumption may be prohibited by legislation following the Montreal Protocol and its successors. The methods selected should be capable of being operated on a reasonably large scale and are required urgently, not least as a discouragement to the illegal traffic in CFCs that has developed.1 It is important that conversion processes do not result in the risk of toxic or ozone-depleting emissions and, ideally, they should lead to useful products. The literature relating to catalytic2 or non-catalytic3 methods for the oxygenation of CFCs is now extensive, the products being CO2 and hydrogen halides. Catalytic hydrogenolysis, for example the conversion of CCl2F2 to the useful, low-temperature refrigerant CH2F2, is also attractive and this reaction is receiving considerable attention.4,5Existing stocks of 1,1,2-trichlorotrifluoroethane held world-wide may be considerable, in view of its former use in a wide variety of industrial applications. In principle, the hydrogenolysis route is applicable here, Scheme 1. Steps i–iii of this scheme form part of the route used in a large scale process for the preparation of CFC-alternative refrigerant CF3CH2F (HFC-134a) from HF + C2Cl4/Cl26 and aspects of the catalytic fluorination7 and hydrogenolysis8 reactions have received fundamental study. As an alternative, CCl2FCClF2 could be converted to CCl2FCF3 and hence to CH2FCF3, via the isomerization CCl2FCClF2 → CCl3CF3 followed by fluorination of CCl3CF3 to CCl2FCF3, steps iv and v of Scheme 1.
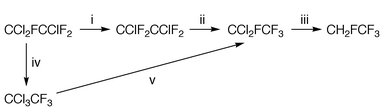 | ||
| Scheme 1 Possible routes from CCl2FCClF2 to CH2FCF3. (i) Fluorination with HF, fluorinated chromia catalyst, refs. 6 and 7. (ii) Isomerization, fluorinated chromia catalyst, refs. 6 and 7. (iii) Hydrogenolysis, Pd/C catalyst, ref. 8. (iv) Isomerization, see text. (v) Fluorination, see text. | ||
Here, attention is focused on the conditions required to achieve facile isomerization of CCl2FCClF2. Mechanistically, this is an interesting reaction. We have previously shown that on heavily fluorinated chromia at 700 K the reaction occurs via an intramolecular process in contrast to other transformations, chlorination or fluorination, that are intermolecular.7
It has been known for many years that CCl2FCClF2 is isomerized extensively to CCl3CF3 when it is refluxed in the presence of aluminium(III) chloride9 and this has been the basis for a large scale batch process to prepare CCl3CF3 as a synthetic intermediate. Extensive chlorination of CCl2FCClF2 also occurs, the stated composition obtained from the reaction under laboratory conditions being CCl3CF3 (50%), CCl3CClF2 (40%), C2Cl6 (5%) and unchanged CCl2FCClF2 (5%). When [36Cl]-labelled aluminium(III) chloride was used, the radiolabel was detected only in CCl3CClF2 and C2Cl6, suggesting that in this case the isomerization is also intramolecular.9 This observation was the starting point for the investigation reported here, undertaken at room temperature under vapour–solid heterogeneous conditions. The original observations have been confirmed but, although isomerization and chlorination are nominally separate processes, the latter is required to produce the catalytically active sites for isomerization. These are believed to be related closely to the recently reported strong Lewis acid aluminium chlorofluoride.10
Results
The behaviour of CCl2FCClF2 vapour over resublimed solid aluminium(III) chloride, aluminium chloride sublimed directly onto calcined γ-alumina and γ-aluminina previously chlorinated with carbon tetrachloride has been studied at room temperature under static, anhydrous conditions. In some instances solids were pretreated with 1,1,1-trichloroethane to determine the effect of a supported organic layer11,12 on the subsequent behaviour of CCl2FCClF2. The components of product mixtures were identified by 19F NMR spectroscopy, GC and GCMS and, although the isomerization of CCl2FCClF2 under the conditions used was slow, its progress could be followed conveniently by quantitative IR spectroscopic measurements made over 24 h periods. Uptakes of CCl2FCClF2 and related CFCs by the solids were determined by mass balance measurements and changes occurring at the surface during series of catalytic reactions by direct Geiger-Müller monitoring13 in experiments where [36Cl]-labelled CCl2FCClF2 or aluminium(III) chloride were used. The key observations made, which must be taken into account in formulating a model to explain the dynamic behaviour of CCl2FCClF2, are summarised below.Mass balance and spectroscopic studies
Solid aluminium(III) chloride and aluminium(III) chloride sublimed directly onto calcined γ-alumina are active catalysts for CCl2FCClF2 isomerization at room temperature, although their activity is reduced markedly by the presence of even trace quantities of water vapour. In contrast, γ-alumina chlorinated with CCl4 shows little or no catalytic ability even though it, like solid aluminium(III) chloride, is a strong Lewis acid capable of room temperature dehydrochlorination of CH3CCl3 and subsequent oligomerization of the CH2![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CCl2 produced.11,12 Exposure of solid aluminium(III) chloride
to CCl2FCClF2 vapour results in an immediate change
in colour, colourless → pale yellow, and the retention of a
substantial fraction of organic material which is removed only with
difficulty by pumping at room temperature. After 24 h exposure, the major
volatile product is CCl3CF3; minor products are the
chlorinated species CCl3CClF2,
CCl3CCl2F and C2Cl6 and traces
of the fluorinated product, CCl2FCF3, are also
present. However the symmetric isomers, CCl2FCCl2F
and CClF2CClF2, were never observed in product
mixtures. Analysis of the retained organic material indicated that
C2Cl6 and CCl3CClF2 were
present. Both compounds have very small vapour pressures at room
temperature (C2Cl6, mp 463–468 K;
CCl3CClF2, mp 314, bp 364 K) and their incomplete
removal by static sublimation in vacuo does not necessarily
indicate any significant interaction involving surface AlIII
centres.
CCl2 produced.11,12 Exposure of solid aluminium(III) chloride
to CCl2FCClF2 vapour results in an immediate change
in colour, colourless → pale yellow, and the retention of a
substantial fraction of organic material which is removed only with
difficulty by pumping at room temperature. After 24 h exposure, the major
volatile product is CCl3CF3; minor products are the
chlorinated species CCl3CClF2,
CCl3CCl2F and C2Cl6 and traces
of the fluorinated product, CCl2FCF3, are also
present. However the symmetric isomers, CCl2FCCl2F
and CClF2CClF2, were never observed in product
mixtures. Analysis of the retained organic material indicated that
C2Cl6 and CCl3CClF2 were
present. Both compounds have very small vapour pressures at room
temperature (C2Cl6, mp 463–468 K;
CCl3CClF2, mp 314, bp 364 K) and their incomplete
removal by static sublimation in vacuo does not necessarily
indicate any significant interaction involving surface AlIII
centres.A similar pattern of behaviour was observed when samples of aluminium(III) chloride, were repeatedly exposed to aliquots of CCl2FCClF2 vapour (up to five) for 24 h periods. The results of two such experiments, including the experimental conditions used, are given in Table 1. In the first experiment complete consumption of CCl2FCClF2 was not achieved, although formation of its isomer, CCl3CF3, was always significant. In the second experiment, the highest conversions to CCl3CF3 were associated with prior addition of a CCl2FCClF2 aliquot from which the formation of highly chlorinated organics had been significant. Most importantly, there was no indication of a chlorine-to-fluorine mass balance, among the organic products, hence a dismutation of CCl2FCClF2 was unlikely. Analogous experiments using aluminium(III) chloride pretreated with CH3CCl3 led to very similar results (Table 2). There is no evidence that the highly unsaturated hydrochlorocarbon oligomers that are the result of the pretreatment,11 inhibit the isomerization of CCl2FCClF2. The purple colour of the oligomer–aluminium(III) chloride surface was discharged during the course of an experiment, the solid becoming grey, and the proportion of highly chlorinated C2 species in the volatile product mixture was somewhat higher (Table 2) than was the case using aluminium(III) chloride (Table 1). Interestingly, pretreatment of aluminium(III) chloride with aliquots of CCl2FCClF2, had no effect on its ability to catalyse CH3CCl3 dehydrochlorination.
| Retained | Volatile materialc (w/w%) | |||
|---|---|---|---|---|
| Addition | materialb | |||
| number | (w/w%) | CCl3CF3 | CCl2FCClF2 | |
| a Reaction conditions: room temperature, 24 h, aluminium(III) chloride (expt. 1, 7.3 mmol, 0.976 g) or (expt. 2, 12.8 mmol, 1.710 g) sublimed directly into the reaction flask in vacuo, CCl2FCClF2, aliquots in the range 4–7 mmol.b Defined as (CCl2FCClF2 − volatile products) g × 100/(CCl2FCClF2) gc Defined as (product) g × 100/(CCl2FCClF2) g, individual components being determined using 19F NMR. Other products observed after each addition were (w/w%): CCl2FCF3 ⩽ 1, CCl3CClF2 ⩽ 3, CCl3CCl2F trace; a build-up of C2Cl6 (identified by GCMS) on the solid was observed in each expt. | ||||
| Expt. | 1 | 25 | 69 | trace |
| 1 | 2 | 0 | 35 | 62 |
| 3 | 5 | 32 | 62 | |
| 4 | 3 | 57 | 37 | |
| Expt. | 1 | 70 | 29 | 0 |
| 2 | 2 | 85 | 13 | trace |
| 3 | 10 | 84 | 0 | |
| 4 | 6 | 89 | 1 | |
| 5 | 48 | 48 | 2 | |
| Retained | Volatile materialc (w/w%) | ||||
|---|---|---|---|---|---|
| Addition | materialb | ||||
| number | (w/w%) | CCl3CF3 | Cl2FCClF2 | CCl3CClF2 | |
| a Reaction conditions: room temperature, 24 h, aluminium(III) chloride (sublimed directly into the reaction flask in vacuo then pretreated with CH3CCl3 vapour for 1 h at room temperature; quantities of aluminium(III) chloride and CH3CCl3 were respectively: 0.01 and 0.03 mol (expt. 1), 0.49 and 0.06 mmol (expt. 2) and 8.7 and 3.9 mmol (expt. 3).b See note b in Table 1.c Defined as in Table 1. Product analyses by 19F NMR (expts.1 and 3) or GC/GCMS (expt. 2). Other products observed after each addition were CCl2FCF3 ⩽ 2, CCl3CCl2F ⩽ 1, C2Cl6 and C2Cl4 (both trace). | |||||
| Expt. 1 | 1 | 45 | 24 | 6 | 6 |
| 2 | 4 | 37 | 53 | 1 | |
| 3 | 3 | 36 | 54 | 3 | |
| 4 | 82 | 15 | trace | trace | |
| 5 | 95 | 0 | 0 | 0 | |
| 6 | 56 | 43 | 0 | 0 | |
| 7 | 98 | 0 | 0 | 0 | |
| Expt. 2 | 1 | 73 | 1 | 0 | 25 |
| 2 | 25 | 67 | trace | 3 | |
| 3 | 38 | 59 | trace | 1 | |
| 4 | 30 | 55 | 0 | 9 | |
| Expt. 3 | 1 | 25 | 72 | 0 | trace |
| 2 | 38 | 57 | trace | 3 | |
| 3 | 37 | 55 | 1 | 2 | |
| 4 | 75 | 17 | trace | 5 | |
The decrease in CCl2FCClF2 in the vapour phase above solid aluminium(III) chloride, aluminium(III) chloride pretreated with one or more aliquots of CCl2FCClF2 or CH3CCl3 or γ-alumina-supported aluminium(III) chloride pretreated with CCl2FCClF2, were inversely related to the formation of CCl3CF3 in the vapour phase as demonstrated by IR spectroscopic measurements over 24 h periods. In all cases three isosbestic points were observed, indicating a direct relationship between consumption of CCl2FCClF2 and formation of CCl3CF3. However, neither of these processes followed simple first or second order kinetics under the conditions used.
The isomerizations of both CCl2FCClF2 and CClF2CClF2 are catalysed by fluorinated chromia at ca. 700 K under flow conditions,7 but there was no evidence that isomerization of CClF2CClF2 occurred at room temperature in the presence of aluminium(III) chloride. No interaction was observed between solid aluminium chloride and CClF2CClF2 or n-C6F14 (which has physical properties similar to CCl2FCClF2) but a mixture of solid CCl3CClF2 and aluminium(III) chloride did react slowly to give a mixture of CCl3CF3, CCl3CCl2F, CCl2FCF3 and CCl2FCClF2. It appears therefore that only chlorofluoroethanes containing CClxF3−x, x = 2 or 3, groups interact with aluminium(III) chloride.
[36Cl]-Radiotracer studies
The extent to which chemical reactions occurred at the surface of aluminium(III) chloride was highly dependent on the degree of hydration/hydroxylation of the surface. For example, the effect of trace H2O, adventitious or deliberately added, on [36Cl] exchange between H36Cl vapour and resublimed aluminium(III) chloride samples is shown in Table 3. The extent to which exchange was observed increased markedly when H2O vapour was deliberately added to the surface prior to H36Cl and was usually measurable when the reagents had been manipulated in a Pyrex vacuum system. It was demonstrated many years ago that no observable [36Cl] exchange occurred between H36Cl and solid aluminium(III) chloride when the materials were rigourously purified.14| Pretreatment of reagents | Fraction of [36Cl] activity exchangeda |
|---|---|
| a Defined as So − St/So − S∞, where So and St are the [36Cl] specific count rates [counts s−1 (mg AgCl)−1] of H36Cl before and after exposure to aluminium(III) chloride; S∞ is the specific count rate calculated on the basis of complete exchange. | |
| H36Cl distilled directly from P4O10 | 0.27, 0.24, 0 |
| None | 0.13, 0.36, 0.55, 0.59 |
| AlCl3 exposed to H2O vapour | 0.60, 0.78, 0.83 |
In contrast, exposure of solid aluminium(III) chloride to [36Cl]-CCl2FCClF2 led to the immediate observation of [36C]-activity on the solid, the count rate due to [36Cl]-CCl2FCClF2 vapour decreasing by 41% over 25 min in agreement with mass balance measurements that indicated 42% retention by the solid. Subsequent addition of aliquots of non-radioactive CCl2FCClF2 had little effect on the surface [36Cl] count rate. [36Cl]-Activity in the vapour phase was too small to quantify precisely but IR spectroscopy showed that CCl3CF3 was the major component. A very slow desorption process occurred over a period of weeks in the counting vessel resulting in a small increase in surface count rate, attributable to a reduction in the β− self absorption effect from the [36Cl]-species on the solid. This was accompanied by the observation of C2Cl6 on the walls of the vessel.
Consistent with these observations, exposure of [36Cl]-labelled solid aluminium(III) chloride to CCl2FCClF2 did not result in the incorporation of [36Cl] into CCl3CF3, formed as a result of isomerization. It is possible that [36Cl] was incorporated to a small extent into the minor, volatile components of the reaction mixture, CF3CCl2F and CCl2FCClF2, but this could not be established definitively.
Exposure of solid aluminium(III) chloride to [36Cl]-CH3CCl3 at room temperature led to a rapid build-up of [36Cl]-activity on the surface of the solid as the purple colour developed. The surface count rate was unaffected by removal of volatile products and fractionation of the latter indicated that H36Cl was formed. Addition of inactive CH3CCl3 to the [36Cl]-labelled purple solid had no effect on the surface count rate, although a small quantity of H36Cl was detected in the vapour. The incorporation of [36Cl] onto the surface when [36Cl]-CH3CCl3 vapour was admitted to an unlabelled purple solid was minimal, indicating that the reaction depends on the availability of aluminium(III) chloride surface sites. Consistent with this view, no [36Cl]-surface count rate and no colour change were observed when aluminium(III) chloride which had been pretreated with H2O vapour was exposed to [36Cl]-CH3CCl3. The reaction was not inhibited if H2O vapour and [36Cl]-CH3CCl3 were added concurrently. These observations, taken together, suggest that H2O can block the adsorption of CH3CCl3 on aluminium(III) chloride but that it does not compete effectively with CH3CCl3, providing the latter is in large excess. The purple layer was hydrolytically unstable. Admission of H2O vapour to the [36Cl]-labelled purple solid resulted in a steady decrease in the surface count rate together with the colour changes, purple → brown → off-white. Some [36Cl] was still retained however.
Admitting successive aliquots of CCl2FCClF2 to aluminium(III) chloride pretreated with [36Cl]-CH3CCl3 as described above, resulted in behaviour which was similar to that observed for aluminium(III) chloride pretreated with [36Cl]-CCl2FCClF2. Significant retention was normally observed and the major components in the vapour above the solid were CCl2FCClF2 and CCl3CF3, although the extent of conversion to the latter was generally less than observed with unlabelled CH3CCl3 (Table 2). [36Cl]-Activity in the vapour was too small to quantify but admission of a CCl2FCClF2 aliquot did have an affect on the [36Cl] surface count rate. An immediate change was observed when CCl2FCClF2 was admitted (increase or decrease observed in different experiments) but thereafter, the surface count rate remained constant over 24 h at its new value. The behaviour indicated that although there was no evidence for [36Cl] incorporation during the isomerization, CCl2FCClF2 → CCl3CF3, changes to the nature of the organic material coating aluminium(III) chloride did occur but in an unpredictable fashion.
The behaviour of CCl4-chlorinated γ-alumina
γ-Alumina chlorinated with CCl4 is a solid Lewis acid which like aluminium(III) chloride is capable of catalysing the room temperature dehydrochlorination of CH3CCl3 and the oligomerization of CH2CCl2 so
formed.12 However mass balance and
spectroscopic data obtained under identical conditions to those in
Table 1 indicated that its activity for
room temperature isomerization of CCl2FCClF2 was very
low. Retention of organic material was observed in all reactions, up to 80%
for extended exposure times. Samples of chlorinated γ-alumina that
had been pretreated with CCl2FCClF2 showed no
dehydrochlorination activity towards CH3CCl3,
indicating that the Lewis acid sites required for dehydrochlorination had
been blocked by the pretreatment.Discussion
The elements of the model proposed to describe the dynamic behaviour of CCl2FCClF2 in the presence of solid aluminium(III) chloride at room temperature can be represented by the following:(a) Adsorption (equation 1)
| CCl2FCClF2 (g) → CCl2FCClF2 (ad) | (1) |
(b) Chlorination (equations 2, 3 and 4)
| CCl2FCClF2 (ad) + AlCl3 (s) → CCl3CClF2 (ad) + ‘AlCl2F’ (s) | (2) |
| CCl3CClF2 + AlCl3 (s) → CCl3CCl2F + ‘AlCl2F’ (s) | (3) |
| CCl3CCl2F + AlCl3 (s) → C2Cl6 (s) + ‘AlCl2F’ (s) | (4) |
Chlorination of C–F bonds by aluminium(III) chloride is well documented9,10,15–17 although such reactions are usually performed above room temperature. The formation of an uncharacterized material, AlFxCl3−x, active for isomerization and dismutation of fluorohalocarbons, was reported15 during a re-examination of the reaction of refluxing CCl2FCClF2 with aluminium(III) chloride. More recently, the very active Lewis acid catalyst, amorphous AlF2.8–2.9Cl0.2–0.1, has been described.10 It is prepared from aluminium(III) chloride and CCl3F in a reaction moderated by CCl4. Calculated fluoride ion affinities of monomolecular aluminium(III) halides, AlCl3−nFn, n = 0–3, are high and from these it has been inferred that amorphous aluminium(III) chlorofluoride has surface sites having similar very high fluoride affinites.10 The material described in equations (2)–(4) as ‘AlCl2F’, although not characterised, may be similar.
(c) Isomerization (equation 5)
| CCl2FCClF2 (g) → CCl2FCClF2 (ad) → CCl3CF3 (g) | (5) |
Consistent with this view is the inhibition of isomerization activity by trace H2O, in agreement with an earlier observation,15 as similar behaviour has been reported for amorphous aluminium(III) chlorofluoride.10
(d) Fluorination
Although fluorination does occur, it is relatively unimportant and was not detected in the initial study.9 Conversion of a C–Cl bond to C–F by Al–F is thermodynamically unfavourable (unlike the analogous CrIII/F/Cl system where the energetics are more balanced) and therefore extensive fluorination is not to be expected.Conclusions
The most obvious way of modifying the catalytic properties of the archetypal solid Lewis acid, aluminium(III) chloride is by treatment with H2O vapour even at a trace level. Experimentally this can be demonstrated by a change in the [36Cl] exchange behaviour of H36Cl toward the solid (Table 3) and can result in complete inhibition of activity towards the room temperature dehydrochlorination of CH3CCl3. It is also likely to be a factor in explaining the degree of irreproducibility observed in the behaviour of CCl2FCClF2 towards aluminium(III) chloride samples (Table 1). More subtle is the modification of the surface that results from chlorination of CCl2FClF2 by aluminium(III) chloride which, we have argued, is required for the catalytic isomerization of CCl2FCClF2 to CCl3CF3 to occur. In this situation aluminium(III) chloride is the catalyst precursor rather than the catalyst and we speculate that the active site for isomerization is a surface AlIII atom in a disordered chlorofluoride environment. In view of the calculated F− ion affinities of molecular binary and mixed aluminium(III) halides,10 it is a moot point whether surface Lewis acidity is promoted by the fluorination process.Somewhat surprisingly, the organic layer, comprising unsaturated
oligomers derived from CH2CCl2 which coats
aluminium(III) chloride as a result of its exposure to
CH3CCl3, has little or no effect on the
CCl2FCClF2 isomerization reaction. The active sites
required are still accessible, presumably due to the quasi-liquid
nature11 of the surface layer and
deactivation by adventitious H2O is still possible.
We have previously used the dehydrochlorination behaviour of CH3CCl3 at an inorganic solid as an operational probe for the Lewis acidity of its surface.12 Using this criterion is was expected that the γ-alumina chlorinated using CCl4 would be active in catalysing the isomerization of CCl2FCClF2 at room temperature. This material would be an attractive alternative to aluminium(III) chloride particularly for large scale use, since it is hydrolytically less sensitive and has a greater surface area. However its isomerization activity is negligible although organic material, which blocks the sites at which CH3CCl3 dehydrochlorination occurs, is retained by the solid. The behaviour of CCl2FCClF2 in the presence of this material differs from that exhibited in the presence of aluminium(III) chloride, either bulk or sublimed onto calcined γ-alumina. A possible reason is that the chlorinating ability of chlorinated γ-alumina is inferior to that of aluminium(III) chloride, presumably for kinetic rather than for thermodynamic reasons. We conclude that the intrinsic Lewis acid strength of surface sites in heterogeneous catalyses involving halocarbon compounds is only one of the factors that must be considered.
Experimental
Standard vacuum and glove box techniques were used throughout. Mass balance experiments were carried out using an evacuable, thin Pyrex bulb equipped with a side arm and designed to enable aluminium(III) to be transferred directly by sublimation in vacuo. Solid transfers were also carried out in a glove box but such samples had lower activity for isomerization. [36Cl]-Radiotracer experiments were performed in a Pyrex counting cell equipped with two intercalibrated Geiger Müller counters, a moveable Pyrex boat which could be positioned under either counter, an ampoule from which a solid could be dropped directly into the boat in vacuo and a gas handling system. The principles that underlie the technique and the application of the method for the study of [36Cl]-halocarbons at inorganic surfaces have been described elsewhere.13,18 Studies of the isomerization of CCl2FCClF2 to CCl3CF3 by IR spectroscopy were carried out in a Pyrex cell fitted with an evacuable ampoule from which solid was dropped into a depression at the bottom of the cell. Windows were either KBr or AgCl. Spectral changes were monitored over 24 h, the cell holder ensuring that positioning of the cell in the spectrometer was reproducible. Instrumentation was: IR, PE 983; 19F NMR, Bruker WP200 or AM200; GC, PE Sigma with FID and PE 8410 with hot-wire detection, 2 m packed column, calibrations being determined experimentally for all compounds; CGMS, VG 7070F.Aluminium(III) chloride was sublimed before use (several times if required) as previously described.11 Calcined γ-alumina (Degussa) was chlorinated with CCl4 at 503 K for 6 h as described elsewhere.18 Samples of resublimed aluminium(III) chloride and chlorinated γ-alumina were treated with CH3CCl3 vapour at room temperature in the mass balance or radiochemical counting cells as appropriate. Both solids behaved as previously reported.11,18
[36Cl]-Labelled compounds were prepared as follows:
CCl2FCClF2 by photolysis (medium pressure Hg lamp,
silica vessel) of a 3∶1 mixture of Cl2 and
CH2FCHF2, CCl4 by the thermal reaction of
a 1∶1 mixture of Cl2 and CHCl3,19 CH3CCl3 by the
iron(III) chloride-catalysed addition of HCl to
CH2CCl2,18
HCl by the reaction of Cl− with conc.
H2SO420 and
Cl2 by KMnO4 oxidation of HCl.21 Solid aluminium(III) chloride was
labelled with [36Cl] by exchange with CCl4 at 60
°C for 24 h.22
Acknowledgements
We thank the EPSRC and ICI for support of this work through CASE awards to D. G. McB. and M. M. McG.References
- See e.g., Chem. Br., Dec. 1998, p. 13; Chem. Eng. News, 1998, Sept. 21st, p. 41..
- See e.g., ; S. Imamura, H. Shimizu, T. Haga, S. Tsuji, K. Utani and M. Watanabe, Ind. Eng. Chem. Res., 1993, 32, 3146 Search PubMed; G. M. Bickle, T. Suzuki and Y. Mitarai, Appl. Catal. B, 1994, 4, 141 CrossRef CAS; E. Kemnitz, A. Kohne and E. Lieske, J. Fluorine Chem., 1997, 81, 197 CrossRef CAS; C. F. Ng, S. Shan and S. Y. Lai, Appl. Catal. B, 1998, 16, 209 CrossRef; S. Tashiro, H. Nagata, M. Kishida, K. Mizuno and K. Wakabayashi, Catal. Today, 1998, 45, 185 CrossRef CAS.
- See e.g., ; H. J. P. de Lijser, R. Louw and P. Mulder, J. Chem. Soc., Perkin Trans. 2, 1994, 135 Search PubMed; J. Burdeniuc and R. H. Crabtree, Science, 1996, 271, 340 RSC; N. Sano, H. Tamon and M. Okazaki, Ind. Eng. Chem. Res., 1998, 37, 1428 CrossRef CAS.
- B. Coq, F. Figuéras, S. Hub and D. Tournigant, J. Phys. Chem., 1995, 99, 11159 CrossRef CAS.
- B. S. Ahn, S. C. Lee, D. J. Moon and B. G. Lee, J. Mol. Catal. A, 1996, 106, 83 CrossRef CAS; E. J. A. X. van de Sandt, A. Wiersma, M. Makkee, H. van Bekkum and J. A. Moulijn, Appl. Catal. A, 1997, 155, 59 CrossRef CAS; A. Wiersma, E. J. A. X. van de Sandt, M. A. den Hollander, H. van Bekkum, M. Makkee and J. A. Moulijn, J. Catal., 1998, 177, 29 CrossRef CAS; E. J. A. X. van de Sandt, A. Wiersma, M. Makkee, H. van Bekkum and J. A. Moulijn, Appl. Catal. A, 1998, 173, 161 CrossRef CAS; M. Makkee, E. J. A. X. van de Sandt, A. Wiersma and J. A. Moulijn, J. Mol. Catal. A, 1998, 134, 191 CrossRef CAS; A. Malinowski, W. Juszczyk, M. Bonarowska, J. Pielaszek and Z. Karpinski, J. Catal., 1998, 177, 153 CrossRef CAS; W. Juszczyk, A. Malinowski and Z. Karpinski, Appl. Catal. A, 1998, 166, 311 CrossRef CAS.
- G. Webb and J. M. Winfield, Chemistry of Waste Minimisation, ed. J. H. Clark, Blackie Academic and Professional, London, 1995, pp. 225–227. Search PubMed.
- L. Rowley, J. Thomson, G. Webb, J. M. Winfield and A. McCulloch, Appl. Catal. A, 1991, 79, 89 CrossRef CAS; and references therein..
- C. Gervasutti, L. Marangoni and W. Marra, J. Fluorine Chem., 1981, 19, 1 CrossRef CAS; Z. Karpinski, K. Early and J. L. d’Itri, J. Catal., 1996, 164, 378 CrossRef CAS; F. H. Ribeiro, C. A. Gerken, G. Rupprechter, G. A. Somorjai, C. S. Kellner, G. W. Coulston, L. E. Manzer and L. Abrams, J. Catal., 1998, 176, 352 CrossRef CAS.
- W. T. Miller Jr., E. W. Fager and P. H. Griswald, J. Am. Chem. Soc., 1950, 72, 705 CrossRef.
- C. G. Krespan and D. A. Dixon, J. Fluorine Chem., 1996, 77, 117 CrossRef CAS; V. A. Petrov, C. G. Krespan and B. F. Smart, J. Fluorine Chem., 1996, 77, 139 CrossRef CAS.
- D. G. McBeth, J. M. Winfield, B. W. Cook and N. Winterton, J. Chem. Soc., Dalton Trans., 1990, 671 RSC.
- J. Thomson, G. Webb and J. M. Winfield, J. Mol. Catal., 1991, 68, 347 CrossRef CAS; J. Thomson, G. Webb, J. M. Winfield, D. Bonniface, C. Shortman and N. Winterton, Appl. Catal. A, 1993, 97, 67 CrossRef CAS.
- J. U. Reid, S. J. Thomson and G. Webb, J. Catal., 1973, 29, 421 CrossRef CAS; A. S. Al-Ammar and G. Webb, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 195 RSC.
- R. L. Richardson and S. W. Benson, J. Am. Chem. Soc., 1951, 73, 5096 CrossRef CAS.
- K. Okuhara, J. Org. Chem., 1978, 43, 2745 CrossRef CAS.
- N. Ishikawa, H. Iwakiri, K. Edamura and S. Kubota, Bull. Chem. Soc. Jpn., 1981, 54, 832 CAS.
- K. C. Eapen, K. J. Eisentraut, M. T. Ryan and C. Tamborski, J. Fluorine Chem., 1986, 31, 405 CrossRef CAS.
- J. Thomson, G. Webb and J. M. Winfield, J. Mol. Catal., 1991, 67, 117 CrossRef CAS.
- H. A. Taylor and W. E. Hanson, J. Chem. Phys., 1939, 7, 48.
- R. N. Maxson, Inorg. Synth., 1939, 1, 147.
- G. Brauer, Handbook of Preparative Inorganic Chemistry, Academic Press, London, 2nd edn., 1965, vol. 1, p. 272. Search PubMed.
- C. H. Wallace and J. E. Willard, J. Am. Chem. Soc., 1950, 72, 5275 CrossRef CAS.
Footnote |
| † Present address: The Leverhulme Centre for Innovative Catalysis, Dept. of Chemistry, The University of Liverpool, Liverpool L69 3BX, UK. |
| This journal is © The Royal Society of Chemistry 2000 |