Nafion resin–silica nanocomposite solid acid catalysts.Microstructure–processing–property correlations
Mark A. Harmer*a, Qun Suna, Alexander J. Vegaa, William E. Farnetha, Alfred Heidekumb and Wolfgang F. Hoelderichb
aDuPont Central Research and Development, Experimental Station, Wilmington, DE 19880-0356, USA.. E-mail: Mark.A.Harmer@usa.dupont.com
bDepartment of Chemical Technology and Heterogeneous
Catalysis, University of Technology RWTH, Aachen, Germany
First published on UnassignedUnassigned8th February 2000
Green ContextNafion is a good example of a solid acid which through structural modification has been made more suitable for organic reaction chemistry. The composites with silica seem particularly promising as replacements for hazardous conventional acids such as hydrogen fluoride. However, like many solid acids, the nature of their activity is poorly understood—especially in terms of distribution of active sites and their local environments. Without this information catalyst optimisation is largely a serendipitous exercise. In this article by one of the pioneers of this type of material, we see how analytical techniques can inform us about the nature of the solid acids and how this information can help to improve catalytic performance.JHC |
Summary
We have developed a new class of solid acid catalyst, a Nafion resin/silica nanocomposite, which effectively catalyzes a wide range of industrially important reactions. These include olefin isomerizations, alkylations, acylations, oligomerizations, the Fries reaction, esterifications and benzylations. We have found that the catalytic activity of these materials is dependent upon the synthetic conditions. The catalytic activity is related to the extent of dispersion of the Nafion resin acid groups within the porous silica framework. The use of an in situ sol-gel technique is preferred over infiltration of a pre-formed support with a Nafion solution. Here we show how we can use solid state NMR, electron microscopy and temperature programmed desorption to elucidate the chemical environment and dispersion of the acid sites. We also show how the microstructure may be tailored depending upon the synthetic conditions. This is a rare example of tuning catalyst activity for particular reactions. In the case of alkylations (such as formation of linear alkyl benzenes), transalkylation and acylation chemistry, the activities are increased several fold using a catalyst in which the Nafion resin is slightly aggregated. A highly dispersed form of the Nafion resin within the silica is preferred for dimerization chemistry. In the case of olefin isomerization chemistry, variations in the Nafion resin dispersion have little effect upon catalytic activity. A correlation of microstructure–processing–properties is described.Introduction
Because of environmental concerns, there has been a strong interest in the use of solid acid catalysts as a replacement to homogeneous catalysts such as HF, AlCl3 and H2SO4.1–10 Although these latter catalysts are very effective, they produce highly corrosive media with chemically reactive waste streams. Purification can be both difficult and hazardous. The major challenge in this area is in the development of cost effective, highly active, selective and stable solid acid replacements. By contrast, the solid acid counterparts are easier to handle, purification is simpler and cheaper, and the general operation of a large chemical process is safer.Over the years, a number of different types of solid catalysts have been developed ranging from sulfated zirconias,1 heteropolyacids,2 zeolites,3,4,11–13 and perfluorinated resinsulfonic acids.5,6 The exact nature of the acid site in these materials and a comprehensive approach to the acid strength remains an area of controversy.7 In the case of sulfated zirconia, long term stability can be a problem due to loss of the active sites whereas in some applications zeolites have high initial activity. Stability however decreases due to the blocking of the small pores by larger product molecules and coke formation. There appears to be a need to fine tune catalyst properties for particular applications. No one single catalyst will be optimal for all acid catalyzed reactions.
Perhaps one of the better characterized solid acid catalysts can be represented by the perfluorinated resinsulfonic acids5,6 which have been reported to have an acidity close to that of 100% sulfuric acid. In particular, Nafion resin which was developed about thirty years ago, is known to catalyze a wide variety of reactions.5,6 Nafion is a copolymer derived from tetrafluoroethylene and perfluoro-2-(fluorosulfonylethoxy)propyl vinyl ether, which after hydrolysis of the sulfonyl fluoride, yields the strongly acidic terminal CF2CF2SO3H group. The equivalent weight of the polymer is typically about 1070 with an acid content of about 0.95 meq g−1. This material is both chemically stable (as expected due to the fluorocarbon nature of the backbone) and thermally stable. Recent reports indicate that Nafion is thermally stable up to 280 °C, at which temperature the sulfonic acid groups begin to decompose.14 One major drawback of the commercially available material (Nafion NR50 resin beads) is the very low surface area (0.02 m2 g−1). The activity of this material either in non-swelling solvents or in the gas phase is very low which in turn has limited the utility of these materials. In order to increase the acid site accessibility of this material we have recently described a new class of solid acid catalyst based upon a high surface area Nafion resin–silica nanocomposite15 where nanometer sized Nafion resin particles are entrapped within a highly porous silica network. This significantly increases the effective surface area of the Nafion particles (within the porous silica) by orders of magnitude and as a result the catalytic activity of this material per unit weight of Nafion resin has been found to be increased up to 1000 times higher than in the pure polymer. A number of very promising applications of this composite material has been described in the literature, ranging from olefin isomerization,16 Freidel–Crafts benzylation,17 dimerization type chemistry,18,19 the Fries rearrangement,20,21 esterifications,22 acylations23 and a number of alkylation reactions.15
We have found that the catalytic activity of these new materials is dependent upon the processing variables, in particular, the source of the silica used. By altering the silica source, we can, in turn, alter the extent of dispersion of the Nafion resin within the porous silica framework. This change of the Nafion–silica microstructure in turn effects the catalytic activity. In the dimerization of α-methylstyrene a highly dispersed form of the Nafion gives the optimum activity. However, in the case of acylation or alkylation chemistry, a slightly aggregated form of the Nafion is preferred. Controlling and understanding the microstructure of these solid acid containing nanocomposites may allow one to fine-tune the characteristics of these materials to optimize both activity and selectivity.
Experimental
Materials. The Nafion resin solution (5 wt% Nafion in a mixed water–alcohol solution) is available from Aldrich. Sodium silicate solutions (20 wt% in silica) and tetramethylorthosilicate [Si(OMe)4] are available from DuPont and Fluka and were used as received. Reagents used in catalysis studies were all of the highest grade available and the liquids were distilled and stored over molecular sieves before use.Catalyst synthesis
The high surface area Nafion resin–silica nanocomposites were prepared according to previously published procedures.15,24 A brief description of the two synthetic methods used is as follows. Both of these methods are based upon an in situ sol-gel technique. In one approach silicon alkoxides were used as the silica source and will be known as the Type 1 catalyst.24 Typically 204 g of Si(OMe)4 were stirred with 33 g of deionized water and 3 g of 0.04 M HCl for 30 mins. In a separate flask, 150 g of 0.4 M NaOH was added to 300 g of the 5 wt% Nafion containing solution. The silica containing solution was added to the basic Nafion containing solution and the whole system gelled to a solid mass in a few seconds. The gel was dried (90 °C, overnight) and re-acidified by repeated washing with 25 wt% nitric acid. Another approach was to use sodium silicate as the silica source. The resulting catalyst will be referred to as Type 2 catalysts. In a typical procedure, 200 g of a 10 wt% silica containing solution (via diluting down a sodium silicate solution) was added to 100 g of a 3 wt% Nafion containing solution and the pH adjusted to 7. Using this approach, the system gelled in about 10–15 s. In all of the catalyst testing, both Type 1 and Type 2 catalysts were fractionated in order to obtain particles of approximately the same size. We were able to alter the pore diameter for the Type 1 materials by adjusting the pH prior to gelation.15 For Type 2, the silicate route, this was achieved by aging the wet gel at temperatures between room temperature and 90 °C, prior to drying. Using this approach, the pore diameter could be varied from about 100 to 200 Å, although these variations did not have a large effect upon the catalytic activity. The Nafion content of both catalysts was verified using both thermogravimetric analysis (using a TGA V5.1 from DuPont) and measuring the acid capacity. Typical Nafion loadings using the above procedure was about 13–14 wt% of Nafion.Catalysis studies
Catalytic reactions were carried out using a batch reactor. In all catalysis experiments, the reactants and solvents were dried for at least 24 h over 4 Å molecular sieves, and the catalysts were dried for 18 h at 150 °C under vacuum. All of the reactions were performed under an atmosphere of nitrogen and the glassware was dried prior to use. The reaction rate was determined from the reactant conversion using only the linear section of the rate curves which essentially represents initial reaction rates. Liquid samples were taken at certain time intervals and separated from the solid catalyst by filtering. All the samples were analyzed by a Hewlett Packard 5890 Series II GC equipped with FID detectors. The product identification was carried out with a GC/MS analysis.For comparative purposes an Amberlyst-15 resin was also used in some experiments. The Amberlyst-15 resin catalyst was washed with an acetone and water mixture several times until a neutral solution was obtained. The washed catalyst has an acid capacity of 4.3 meqH g−1. Amberlyst-15 resin was dried at 110 °C. Benzene and 1-dodecene were purchased from Aldrich. Both reagents were distilled and 3 Å molecular sieves were added to keep the reagents dry. The purity of the 1-dodecene was 95%, which contained ca. 4% branched olefins.
Characterization
BET surface area, pore diameter, and BJH cumulative pore volume were obtained using a Micromeritics instrument. Scanning electron microscopy (SEM) was performed using a Hitachi S-5000SP scanning electron microscope.Results and discussion
(I) Alkoxide based in situ Nafion–silica composites
We have developed Nafion resin–silica nanocomposites, made by an in situ sol-gel technique.15 A detailed description of the silica chemistry can be found in the literature.25 Soluble silica precursors, such as silicates or alkoxy silanes, were mixed with a nanometer-sized colloidal dispersion of Nafion in a polar solvent. As the gel was dried, the Nafion based particles became entrapped within the network as it was formed. Fig. 1 shows a schematic of how the composite was formed. The silica network forms via the aggregation of nanometer sized silica particles. The gel shrinks during this condensation stage. During the final drying, shrinkage is accelerated due to capillary forces which results in further condensation. A porous continuous network results with a very high surface area. The Nafion particles become entrapped within this porous network. The Nafion is readily accessible due to the pores in the silica network which are in the range of 10–25 nm. The final Nafion resin–silica nanocomposite is shown in Fig. 2.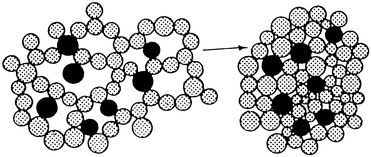 | ||
| Fig. 1 The Nafion resin particles (●) become entrapped within the structure as the silica particles condense to form a network of porous silica gel. | ||
 | ||
| Fig. 2 Nafion resin–silica nanocomposite. | ||
The surface area of these new nanocomposites is approximately 20000 times greater than that of the starting polymer, with surface area typically 150–500 m2 g−1. Variations of the synthesis can be used to alter pore structure, surface area and acid loading in order to optimize activity. The Nafion resin is dispersed throughout the porous silica network. As shown previously using energy dispersive X-ray analysis for elemental F (from the Nafion) and Si (from the silica) we have shown the two materials are intimately mixed at the nanometer level.15
(II) Dispersed Nafion catalysts on pre-formed supports
The in situ approach has many advantages compared to attempts to disperse the Nafion resin upon a pre-formed support. One alternative to the in situ method is to take a pre-formed high surface area silica support and deposit the Nafion over the entire support. We have found however, this approach has several drawbacks. Firstly, the Nafion resin solutions are not really true solutions, but a fine colloidal dispersion and the polymer does not significantly penetrate within the internal pores of the support, but sits as larger micron sized lumps on the outer surface of the support. Fig. 3 shows SEMs (including X-ray image analysis) resulting from the two synthetic approaches. We have used a number of pre-formed silica supports and the results were essentially the same for all of these. The support surface area was typically in the range of about 200–400 m2 g−1, with a pore diameter around 15 nm (for example using a Grace Davison 62 and Grace Davison 653 silica support). The highly dispersed and entrapped Nafion resin–silica nanocomposite via the in situ approach is unattainable using the pre-formed support. In this latter case the Nafion resin does not coat the bulk on the internal surface but rather coats the very low external surface area resulting in a film in the micron sized range (the coating is also non-uniform and some regions have no Nafion). Using this latter approach, a much poorer dispersion is obtained. | ||
| Fig. 3 SEM of an in situ developed Nafion resin–silica nanocomposite showing the Si map (top, center) and F map (top, right). Below shows an SEM of a material prepared using a pre-formed support (same scale), showing the Si map (center) and F map (right); the Nafion does not penetrate into the support but simply sits on the very outer surface. | ||
In addition to giving a much improved polymer dispersion the in situ approach leads to a more stable material with respect to solvent leaching. The nanocomposite can be stirred in refluxing ethanol for 48 h with no leaching of the Nafion (from TGA analysis and by measuring the number of acid equivalents), whereas using the pre-formed support, in excess of 40 wt% is washed off over the same period. The catalytic activity is also higher for the in situ nanocomposite versus using a pre-formed support. For the nanocomposite, the heptene alkylation of toluene and the 1-dodecene alkylation of benzene over a period of about 2 h gives conversons in the range of 90–95%. This is compared to about 25–50% for the pre-formed support. A number of other supports were also investigated (a range of low and high surface area silicas, some low surface area aluminas, some high porosity PTFE based supports such as Chromosorb T). In all cases, lower activities were found compared to the in situ method sol-gel method. In summary, the in situ approach appears to have the advantages of improved dispersion, higher effective surface area of the Nafion and improved stability, all of which lead to major improvements in catalytic performance.
(III) Tailored Nafion–silica composites based upon in situ sol-gel techniques: characterization
We have also recently found that we can tailor the microstructure of the in situ prepared Nafion resin–silica nanocomposites. An initial report of this has appeared in the literature.24 Small variations within the synthesis (mainly the nature of the silica network precursor) leads to pronounced changes in the catalytic activity. In going from an silicon alkoxide precursor (Type 1) to a silicate type precursor (Type 2), has in general led to an increase in catalytic activity compared to results reported earlier.15Possible differences between the Type 1 and Type 2 catalysts can be elucidated using pore structure analysis, microscopy and temperature programmed desorption. In an earlier study, we suggested the Nafion particle size was in the range of 20–60 nm for the alkoxide source material. We examined the surface area, pore size, pore size distribution and pore volume of calcined composites (600 °C at which temperature the polymer is completely removed) and compared the before (polymer present) and after (polymer absent) microstructures. In the case of the Type 1 material the major changes in the pore structure occur in the 50 to 300 Å range. For Type 1, the nitrogen adsorption isotherms indicated that upon calcination the average pore diameter had increased to about 216 from 146 Å (the pore volume also went up to about 0.95 from about 0.75 cm3 g−1). Careful examination of the data indicates the Nafion particle size is in the range of about 5–30 nm (a bit lower than previously reported) with the majority in the range of about 10–20 nm. Before calcination, the porosity had an envelope in the range of about 80–250 Å with an average pore size of 146 Å. Upon calcination, the general envelope of pore size distribution (pore diameter for a particular pore volume) shifted to higher values. Control experiments with silica itself did not show any appreciable change after calcination to 600 °C. The increase in pore size and pore volume is consistent with the model that the Nafion is distributed throughout the silica at the 10–20 nm level.
In contrast, for the Type 2 material, only a small change (5%) occurred in the pore structure in 0–600 Å pore diameter range. The average pore diameter before calcination was about 138 Å and this shifted slightly to 149 Å (measured using nitrogen, BJH). The major change for the type 2 materials however occurred in the sub-micron range with a new collection of pores found in the 0.1 to 0.25 micron range using mercury intrusion data. The change over this region was not observed for the Type 1 materials. In this case, the data is consistent with the Nafion being dispersed throughout the silica at the 0.1 to 0.25 micron level with a much smaller population at the 10–20 nm level.
The difference between the two materials can also be seen in Fig. 4, using scanning electron microscopy. Fig. 4(a) shows the SEM (polished cross section) of the Type 1 Nafion resin–silica catalyst material, where the Nafion has been removed at 600 °C via calcination. We have used this approach before to highlight where the Nafion resided within the silica microstructure. As can be seen in Fig. 4(a), only small pores (consistent with our estimate above of <30 nm sized particles) are evident. In Fig. 4(b) (for the Type 2 material) a new collection of pores on a larger scale (in the 0.1 to 0.2 μm range) is found. This is consistent with at least some of the Nafion particles aggregating into larger particles within the porous silica network. In all of these experiments several areas were investigated with similar findings.
![SEM micrographs of Nafion resin–silica composites [Type
1(a) and Type 2(b)] after calcination to 600 °C.](/image/article/2000/GC/a907892d/a907892d-f4.gif) | ||
| Fig. 4 SEM micrographs of Nafion resin–silica composites [Type 1(a) and Type 2(b)] after calcination to 600 °C. | ||
The microscopy and pore size data are consistent with the Nafion resin in the Type 2 catalyst being more aggregated than in Type 1. This data is also consistent with some of the experimental observations during synthesis of these material. It was noticed that upon mixing the Nafion solution with sodium silicate (without the addition of acid to induce gelation) that the solution became slightly cloudy within a few seconds. When the solution was left overnight then the Nafion polymer settled out, above which was a clear silica containing layer. Such a settling was not observed upon mixing hydrolyzed alkoxide pre-cursors with the Nafion solution. It appears that the sodium silicate solution (with its very high ionic strength at pH 12–13) induces some slight aggregation of the Nafion polymer in which case it may be better to describe the Type 1 catalysts as ‘dispersed’ compared to the Type 2 catalysts.
Supporting evidence for this more aggregated Nafion resin dispersion comes from TGA/TPD experiments (adsorption/temperature programmed desorption) using isopropanol as the reactant. Essentially complete conversion to propylene and other hydrocarbons (secondary products) occurs on all three materials. The high temperature peak in the desorption from NR50 (and to a lesser extent the Type 2 composite) is mostly fragments of higher hydrocarbons, not propylene. Only on the Type 2 catalysts is there a very small amount of propanol evolved. At the same Nafion loading, the rate of adsorption of isopropanol within the sodium silicate derived composites, occurs at a much lower rate than using the alkoxide based materials. This is consistent with a larger Nafion domain size in the former case where the isopropanol uptake rates are more like that of pure Nafion. The desorption characteristics of the Type 2 catalysts are also more ‘Nafion-like.’ This is reflected in the broader desorption traces for primary products (propylene and water) and the appearance of a significant amount of secondary product formed for these materials, Fig. 5. The product desorption peaks for the Type 1 composite is complete at 150 °C. The desorption behavior of the pure polymer is not clean and extensive desorption still takes place at 200 °C due to the poor diffusion. The Type 2 material behavior is somewhat in between the two, which is consistent with a larger domain size of the Nafion within the Type 2 materials.
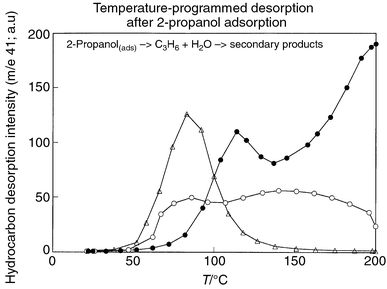 | ||
| Fig. 5 Temperature-programmed desorption after 2-propanol adsorption on Nafion NR50 (●), Type 2 composite (○) and Type 1 (△). The ramp rate was 5 °C min−1. | ||
The microstructure of the two respective catalysts has been investigated using solid state NMR. For comparison the NMR of the pure polymer Nafion NR50 was also studied. Fig. 6 shows the proton MAS spectra of the 13 wt% Nafion–silica composites (both Type 1 and Type 2) upon gradual exposure to water vapor.
 | ||
| Fig. 6 Proton MAS spectra of 13% Nafion–resin silica composites shown for the Type 1 and Type 2 material as a function of gradual exposure to water vapor. The proton MAS NMR of Nafion is shown for comparison (top right). | ||
The interaction of the Nafion with water using MAS NMR at room temperature has been reported by Fraissard et al.26 The chemical shift of the unsolvated hydronium ion in Nafion with one water molecule per acid site, determined by 1H MAS NMR at room temperature, was reported to be 10.4 ppm relative to external TMS. Upon increasing the water content (up to 2.1 water molecules per acid site), the chemical shift was found to decrease to 8.5 ppm. This is also in agreement with earlier studies where it has been found that the proton NMR of the Nafion decreases continuously with increasing water content. The peaks around 5 ppm and below are due to the silica network (silanols for example) and shall not be discussed further. These peaks are obtained when the synthesis is carried out in the absence of the Nafion. There is a clear difference between the Type 1 and Type 2 microstructure. In the case of the silicate derived material (Type 2) the proton NMR is characteristic of a composite of Nafion and silica. The chemical shift of the unsolvated hydronium ion in Nafion is present as a broad band around 10.4 ppm (in agreement with Nafion, ref. 26). As the water level increases, this peak becomes sharper and shifts to a smaller chemical shift (a value of 8.5 ppm at 5H per sulfonate group). The peak position was in agreement with the proton NMR of bulk Nafion which shows a sharp peak at 8.6 ppm. In contrast, no peaks due to the hydronium ion of the Nafion, could be detected in the Type 1 material. The only signals observed correspond to shifts in the silicate spectrum. Although the exact reason for the apparent absence of the Nafion peak is not known, one possible explanation is that the acid sites are well dispersed on and within the silica surface. This may, in turn, lead to considerable line broadening. Consistent with the TPD data above the microstructure in the Type 2 composite is more ‘Nafion like’ than in the Type 1 composite.
(IV) Tailored Nafion–silica composites based upon in situ sol-gel techniques: catalysis
Table 1 shows a comparison of a number of catalytic reactions investigated using the Nafion resin–silica nanocomposites prepared from the two alternative silica precursors. In general, using sodium silicate as the silicate source (Type 2), leads to a Nafion resin–silica microstructure which is about 10 times more active in the industrially important area of alkylations than previously reported using the alkoxide route (Type 1),15 specifically in the formation of cumene and linear alkyl benzenes, Table 1. Using the synthetic conditions as described above leads to materials with very similar pore structures. Typical values of surface area, pore volume and pore size for the Type 1 and Type 2 catalysts are 201 (m2 g−1), 0.77 (cm3 g−1), 146 Å and 165 (m2 g−1), 0.65 (cm3 g−1), 127 Å respectively. The Nafion content of both of these materials was in the range of 13–14 wt% (determined using TGA) and the number of acid sites ca. 0.15 meq g−1 as described previously.15| Rate [mM/(meqH+ h)] | |||
|---|---|---|---|
| Reaction | Type 1 (via alkoxide route) | Type 2 (via silicate route) | Conversiona (%) |
| a Conversions shown are found using the optimum amount of catalyst (typically 5 wt% loading) according to the following conditions: (a) At 80 °C from benzene (21.5 g) alkylation with 1-dodecene (10 g) using 2 g of catalyst, 2 h, (b) acylation of m-xylene (21 g) with benzoyl chloride (10 g) at 140 °C, 2 h, 1 g of catalyst; (c) benzene alkylation with propene at 70 °C (using a 20 g solution of benzene saturated in bubbling propene) with 1 g catalyst, 1 h; (d) tert-butyl-p-cresol (10 g) with toluene (30 g) with 1 g of catalyst at 115 °C, 1 h; (e) 1-dodecene (25 g) at 80 °C with 1 g of catalyst, 1 h; (f) α-methylstyrene dimerization (6 g of AMS in 54 g of cumene as solvent, 0.5 g of catalyst added) at 50 °C, 20 min; (g) decomposition of cumene hydroperoxide (35 g) with 0.1 g catalyst at 50 °C to form phenol and acetone, 1 h. | |||
| Benzene/dodecene alkylation (a) | 32 | 430 | 99 |
| Transalkylation (d) | 1100 | 3086 | 98 |
| Benzene propylation (c) | 46 | 289 | 30 |
| Freidel–Crafts acylation (b) | 200 | 900 | 80 |
| 1-Dodecene isomerization (e) | 1280 | 1580 | 99 |
| AMS dimerization (f) | 6500 | 1980 | 98 |
| Hydroperoxide decomposition (g) | 1750 | 1040 | 99 |
Table 1 shows the catalytic activity using the Type 1 and Type 2 catalyst for seven different reactions including alkylations, acylation, olefin isomerization, the dimerization of α-methylstyrene and hydroperoxide decomposition. Although both catalysts have a similar pore structure and Nafion loading the activity varies quite dramatically. What is particularly interesting is whereas catalyst Type 2 is very effective for alkylation chemistry, the reverse is true in the case of the AMS dimerization and hydroperoxide decomposition. In the case of olefin isomerization, there appears to be only a small difference between the two materials investigated. For each reaction, the catalyst and conditions have been optimized to give very high conversions with good selectivity. The selectivity did not vary appreciably for the two different types of catalysts. As shown below, for example, the two catalysts gave about 28% of 2-dodecyl-substituted benzene. In the acylation type chemistry, the main acylated product (ca. 85%) occurs at the 4-ring position using 1,3-xylene, with minor amounts of the 5-ring (meta to the CH3, ca. 1%) and about 13% in the 2-ring position. In the case of α-methylstryrene chemistry, the only three products observed were 2,4-diphenyl-4-methyl-1-pentene (I), 2,4-diphenyl-4-methyl-2-pentene (II) and the saturated dimer 1,1,3-trimethyl-3-phenylindan (III).18 No higher oligomers were found. As shown in reference 18, typical selectivities were about 72% (I), 16% (II) and 12% (III). Similar values were obtained using either the Type 1 or Type 2 materials.
As shown above, we have been able to tune the catalytic activity of a number of industrially important reactions by tailoring the microstructure. A more detailed account of two of these reactions is described below. Improvements in catalytic activity have been found in a number of areas, for example in the area of alkylations and transalkylations. A good example of this was observed in the use of these catalysts in the formation of linear alkylbenzenes. Linear alkylbenzenes (LAB) are used in the production of linear alkylbenzene sulfonates (LAS), which are extensively used as surfactants. Over 2 million metric tons of LAB are produced each year. The total market for LAS exceeds $30 billion. Currently, it has primarily been produced by the HF catalyzed alkylation of benzene with C10–C16 olefins and the AlCl3 catalyzed alkylation of benzene with chloroparaffins.27 The products of these reactions contain a mixture of alkylbenzenes with the phenyl group attached to different C-atoms in the linear hydrocarbon chain. The 2-phenyl isomer is the most preferred product.

Branched isomers, which are a result of skeletal isomerization of the linear hydrocarbon chain, are very undesirable due to lower biodegradability. This leads to extensive environmental problems.28
Owing to the corrosive nature of mineral acids, solid acid catalysts have been sought for the production of LAB. Various solid acids including zeolites,29,30 heteropolyacids31 and clays32,33 have been investigated for LAB formation via alkylation of benzene with linear olefins. Recently, UOP has developed a new process using a fluorided silica-alumina catalyst.28 It was indicated that this process has two advantages over the HF process. It produces LAB with higher 2-phenyl isomer content (28% vs. 17% from HF in average) and higher linearity (94.5% vs. 93% from HF in average).
In this work we have studied LAB formation by using the alkylation of benzene with 1-dodecene. Both composite catalysts were tested. The composite catalysts not only show very high activity but also produced LAB with very high linearity (>99%). The formations of linear alkyl benzenes (LAB) via the alkylation of benzene with 1-dodecene are listed in Table 1 for the Type 1 and 2 catalysts. With 2 g of the composite catalyst (Type 2), 99% dodecene conversion was obtained after 2 h at 80 °C, with an activity based upon initial reaction rates about 10× that of the Type 1 composites. At 99% dodecene conversion, the products contain >95% LAB and the remainder (<5%) are the ca. 4% branched alkylates from the ca. 4% branched olefins (impurity in the feed), dimers of 1-dodecene and disubstituted benzene. The linearity of the alkylation using 1-dodecene is >99%. The alkylation product distribution (among the LAB) changes with the benzene conversion. In general, at low conversion more 2-phenyldodecane is obtained. Values for the LAB product distribution are as follows: 2-Ø(28%), 3-Ø(19%), 4-Ø(16%), 5-Ø(17%) and 6-Ø(20%) where Ø denotes phenyl and the number refers to the substitution on the dodecene.
The LAB product distribution is quite typical from solid acid catalysts34,35 including a mechanistic study by Beck et al.35 who have shown the influence of transition state selectivity and/or pore diffusion on the isomer distribution. The 28% 2-phenyldodecane selectivity is significantly higher than that obtained from the HF process (15–18%). In addition to the high activity (high conversion) the major advantage of the Type 2 catalyst is the very low production of the undesirable branched alkylbenzenes, of which even in the UOP process ca. 5–7% are produced. This is primarily due to the fact that, unlike HF and AlCl3, the Nafion–silica catalyst does not readily catalyze the skeletal isomerizations of linear olefins.
Interestingly, we have also found that the activity of the Type 2 catalysts was approximately 400× greater than an Amberlyst-15 and pure Nafion (Nafion NR50) catalyst (same weight of catalyst). Typical values of conversion (%) and initial reaction rate (mM/meqH+ h) for the composite, Amberyst-15 and Nafion NR50 are 90(430), 4(1) and 1(1) respectively. In comparison to the pure polymer these results illustrate the greater accessibility to the strong acid sites within the high surface area composite (compared to the non-porous, non-swollen pure Nafion polymer) in this reaction medium. In the case of the Amberlyst catalyst these results presumable reflect the inherent higher acid strength of the perfluorosulfonic acids sites as compared to the phenyl sulfonic acids.
In conclusion, sodium silicate derived 13 wt% Nafion resin–silica composite is very active for catalyzing LAB formation via the alkylation of benzene with linear olefins. Very high linearity (>99%) and higher 2-phenyldodecane selectivity are obtained compared to the current homogeneous acid catalyzed processes.
We have also found higher activities using the Type 2 catalysts in acylation and transalkylation chemistry. The transalkylation between 2-tert-butyl-p-cresol and toluene was studied.

Substituted phenols and derivatives are widely used in polymer resins, agricultural products, and antioxidants in fuels and fragrances. Transalkylation is a significantly more demanding chemistry than the alkylation of phenol with olefins, typically requiring strong acid catalysts and elevated temperatures. The reaction rate from the Nafion resin–silica (Type 2) was around 3 times higher than that from Type 1, with conversions of 99.6%. It appears that more aggregated Nafion resin particles in the composite may lead to higher activity for these more demanding chemistries in general. Comparisons of the Type 2 catalysts with Nafion and Amberlyst-15 also revealed higher activities. Typical values of conversion (%) and initial reaction rate (mM/meqH+ h) for the composite, Amberyst-15 and Nafion NR50 are 99.6 (463), 87 (79) and 55 (27). Some swelling of the pure Nafion polymer does occur in these types of phenolic resins giving rise to modest activity although the improved dispersion within the composite leads to almost quantitative conversion. In both of the above reactions the use of an highly active solid acid catalyst will have a number of processing advantages over the homogeneous analogues. We have also found that these catalysts can be regenerated in most cases. As we have reported previously24 for some reactions such as the isomerization of olefins, these materials exhibit excellent lifetime stability with greater than 10000 turnovers. After this period the catalyst begins to loose activity and darkens in colour (brown). The catalysts can be regenerated using nitric acid (using 25 wt% HNO3 at 50 °C for 4 h). We have restricted these studies to non-aromatic based reactants due to potential reactions of the aromatics with the nitric acid.
It is interesting to rationalize the microstructural causes for the activity patterns shown in Table 1. However, each one of these reactions has a unique structure–activity profile that ultimately derives from the combined effect of microstructure and a complex sequence of elementary kinetic steps that is different for each reaction type. We know very little about these mechanisms, so our observations about the underlying effect of microstructure is speculation, at best. With that disclaimer, here are some observations. The reactions that are faster for the Type 1 catalyst are those that are the most likely to be rate-limited by the initial proton transfer. We have demonstrated that proton transfer is rate-limiting for the AMS dimerization.18 This is probably the case for the peroxide decomposition as well, where the protonated hydroperoxide–sulfonate anion pair should not be unstable. It seems logical that the reaction rates are optimized for the catalysts that have the better dispersed, more accessible acid site distribution when proton transfer is rate-limiting. On the other hand, reactions (a)–(e) probably all involve discrete intermediates in the rate limiting step. Reactions (a), (c) and (e) presumably proceed through protonated olefin intermediates, which are likely present under the reaction conditions, primarily as covalently bound sulfonate esters. We have evidence that the chemical rate limiting step in the 1-dodecene isomerization, (e), is ester decomposition. Similarly (a) and (c) are likely to proceed through sulfonate esters, and a key feature of the ester reactivity as an electrophile for aromatic substitution may be the energetic difference between its neutral and ion pair forms. The relative stabilities of neutral and ion pair structures will be sensitive to the microenvironment, a higher effective dielectric constant, for example, or a more fluid local ‘solvation sphere’, favoring ion pair stabilization. We speculate that the more ‘Nafion-like’ acid site environments of the Type 2 catalysts can enhance the rates of reactions like (a)–(e) through these types of medium effects on intermediate and transition state stabilities. In the case of the Type 1 materials we have modified the silanol groups (controlled reaction of these composites with HF vapor using an HF/pyridine mixture) to convert some of the Si–OH groups to Si–F groups. Treating materials prepared using the alkoxides leads to a five fold increase in alkylation activity. Again this reflects the importance of micro environments upon activity.
Two interesting reports by Palinko et al.36 and Botella et al.37 appear consistent with some of our findings. Palinko et al.36 have reported on the surface characterization of both Nafion and a Nafion–silica composite catalyst by infrared microscopy. The composite was made using silicon alkoxide and would correspond in our study to a Type 1 catalyst. One conclusion reached was that of an interaction of the highly dispersed sulfonate groups SO3H containing pockets of the Nafion and the hydroxyl groups of the silica within the composite. It was also suggested that this may lead to some decrease in acidity. The influence of the composition within the Nafion–silica composites on the isobutane/2-butene alkylation has also been reported.37 A similar conclusion was also observed. In the highly dispersed Nafion in silica systems (made from silicon alkoxides), the sulfonic groups of the polymer interact to a greater extent with the silanol groups of the silica, resulting in a decrease in the activity of the sulfonic acid groups. In the Type 2 materials, where the Nafion is more aggregated, one would expect the interaction of the sulfonate groups (SO3H) and the hydroxy groups (SiOH) of the silica to be considerably reduced, leading to a material with an acidity which is more ‘Nafion-like’. A more detailed study is required to fully understand all of these effects. Overall, then, we are inclined to think of the ratio of rates for Type 1 and Type 2 catalysts as the result of a trade-off between acid site accessibility, more favorable for Type 1 catalysts, and acid site ion pair micro-solvation effects, which may have a higher inherent acidity, more favorable for Type 2 catalysts. The observed activity is a complex function of the reaction mechanism and reaction conditions.
Summary
We have described a number of industrially important reactions that can be catalyzed by Nafion resin–silica nanocomposites. The activity of the catalyst is very dependent upon the processing. An in situ sol-gel method gives a catalyst with the highest activity. The sol-gel method can also be adapted to alter the dispersion of the Nafion within the porous silica network. This in turn has allowed fine tuning of catalyst activity. This kind of material may represent a very attractive alternative for the more hazardous homogeneous acids such as triflic acid, HF, BF3 and aluminium trichloride.Acknowledgements
A. H. and W. F. H. are grateful to financial support of DuPont. We are also grateful to Mr Steven Winchester for excellent technical support and Mr Dennis Smith for help with electron microscopy.References
- X. Song and A. Sayari, Catal. Rev. Sci. Eng., 1996, 38, 329 Search PubMed.
- I. V. Kozhevnikov, Catal. Rev. Sci. Eng., 1995, 37, 311 Search PubMed.
- P. B. Venuto, Microporous Mater., 1994, 2, 297 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
- G. A. Olah, S. I. Pradeep and G. K. S. Prakash, Synthesis, 1986, 513 CrossRef CAS.
- G. A. Olah, in Acidity and Basicity of Solids, ed. J. Fraissard and L. Petrakis, Kluwer, Dordrecht, Netherlands, 1994, pp. 305–334. Search PubMed.
- T.-K. Cheung and B. C. Gates, Chemtech., 1997, 28 Search PubMed.
- W. F. Hoelderich, Stud. Surf. Sci. Catal., 1993, 75, 127 Search PubMed.
- W. F. Hoelderich and E. Heitmann, Catal. Today, 1997, 38, 227 CrossRef CAS.
- W. F. Hoelderich and D. Heinz, Catal. Today, 1999, 14, 149.
- W. F. Hoelderich and H. VanBekkum, Stud. Surf. Sci. Catal., 1991, 58, 631 Search PubMed.
- W. F. Hoelderich, in Comprehensive Supramolecular Chemistry, ed. A. Guilio and T. Bein, 1996, vol. 7, ch. 23, Part II, Elsevier, England, pp. 671–682. Search PubMed.
- P. Espel, R. Parton, H. Toufar, J. Martens, W. F. Hoelderich and P. A. Jacobs, in Catalysis in Zeolites, ed. J. Weitkamp and L. Pupper, 1999, Springer, Heidelberg, pp. 377–436. Search PubMed.
- S. R. Samms, S. Wasmus and R. F. Savinell, J. Electrochem. Soc., 1996, 143, 1498 CAS.
- M. A. Harmer, W. E. Farneth and Q. Sun, J. Am. Chem. Soc., 1996, 118, 7708 CrossRef CAS.
- Q. Sun, M. A. Harmer and W. E. Farneth, Chem. Commun., 1996, 1201 RSC.
- Q. Sun, M. A. Harmer and W. E. Farneth, Ind. Eng. Chem. Res., 1997, 36, 5541 CrossRef CAS.
- Q. Sun, W. E. Farneth and M. A. Harmer, J. Catal., 1996, 164, 62 CrossRef CAS.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, Catal. Lett., 1997, 47, 243 Search PubMed.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, ACS Polym. Prepr., 1997, 763 Search PubMed.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, J. Catal., 1998, 176, 260 CrossRef CAS.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, J. Catal., 1999, 181, 217 CrossRef CAS.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, J. Catal., 1999, 188, 230 CrossRef CAS.
- M. A. Harmer, W. E. Farneth and Q. Sun, Adv. Mater., 1998, 10, 1255 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, in Sol-Gel Science, Academic Press, 1990. Search PubMed.
- P. Batamack and J. Fraissard, Catal. Lett., 1995, 35, 135 Search PubMed.
- Linear Alkylbenzenes, 93S10, Chem. Systems, August, 1995. Search PubMed.
- J. A. Kocal, US Pat., 5,344,997, 1994..
- S. Sivasanker and A. Thangaraj, J. Catal., 1992, 138, 386 CrossRef CAS.
- L. B. Young, US Pat., 4,301,317, 1992..
- R. T. Sebulsky and A. M. Henke, Ind. Eng. Chem. Process Des. Dev., 1971, 10, 272 Search PubMed.
- M.-Y. He, L. Zhonghui and M. Enze, Catal. Today, 1988, 2, 321 CrossRef CAS.
- T. J. L. Berna and D. A. Moreno, Eur. Pat. Appl., EP 0353,813, 1981..
- S. Sivasanker, Proceedings 10th International Congress on Catalysis, Elsevier, Amsterdam, 1992, p. 397; US Pat., 4329509..
- Handbook of Heterogeneous Catalysis, ed. G. Ertl and H. Knoezinger, Wiley-VCH, 1997, vol. 5, p. 2130. Search PubMed.
- I. Palinko, B. Tork, G. K. S. Prakash and G. Olah, Appl. Catal., 1999, 174, 147 CrossRef CAS.
- P. Botella, A. Corma and J. M. Lopez-Nieto, J. Catal., 1999, 185, 371 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |