Fluorescence of solvent-complexed aminobenzonitriles in a supersonic jet
Oliver
Krauß
a,
Uwe
Lommatzsch†
a,
Christoph
Lahmann
a,
Bernhard
Brutschy
*a,
Wolfgang
Rettig
b and
Jerzy
Herbich
c
aJohann Wolfgang Goethe-Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersität, Marie-Curie-Str. 11, D-60439, Frankfurt am Main, Germany. E-mail: brutschy@chemie.uni-frankfurt.de
ersität, Marie-Curie-Str. 11, D-60439, Frankfurt am Main, Germany. E-mail: brutschy@chemie.uni-frankfurt.de
bHumboldt-Uniersität, Bunsenstr. 1, D-10117, Berlin, Germany. E-mail: rettig@chemie.hu-berlin.de
cInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland. E-mail: herbich@ichf.edu.pl
First published on 5th December 2000
Abstract
The fluorescence of aminobenzonitrile clusters with solvent molecules under supersonic jet conditions is studied. The cluster size distribution is determined by time-of-flight mass spectrometry with an experimental setup that allows for mass analysis and dispersed emission spectroscopy quasi-simultaneously. For 4-dimethylaminobenzonitrile (DMABN) dual fluorescence is observed for the monomer when complexed with a minimum of five acetonitrile molecules. The single emission bands of the 4-aminobenzonitrile (ABN) and 1-ethyl-2,3-dihydro-indole-5-carbonitrile (EIN) solvent complexes are red-shifted with respect to the uncomplexed chromophores. The difference between DMABN and ABN/EIN is attributed to the population of the CT state in DMABN.
1. Introduction
In 1959 Lippert discovered that the emission spectrum of 4-dimethylaminobenzonitrile (DMABN) in polar solvents is composed of a short and a long wavelength emission band (dual fluorescence phenomenon).1 The presently most widely accepted explanation of this behaviour has been proposed by Grabowski et al.2,3 and reviewed by Rettig4 in terms of the twisted intramolecular charge transfer (TICT) model. This model assumes that the unusual red-shifted emission band is due to an intramolecular electron transfer (ET) from the dimethylamino group to the benzonitrile moiety. The ET is accompanied by a twist of the donor group that leads to an orthogonal conformation of the dimethylamino group with respect to the phenyl ring. The TICT model has been criticized by several authors offering alternative explanations. Zachariasse et al.5 proposed the amino inversion mode and the S1/S2 energy gap as the decisive parameters in the phenomenon, while Visser and Varma6 suggested another mechanism related to a solute–solvent exciplex formation.Excited state ET is a fundamental and complex phenomenon playing a crucial role in a variety of photophysical, photochemical and biochemical processes. DMABN and related compounds are widely used for studying the structural requirements, the thermodynamics, the kinetics and the solvation effects of photoinduced ET. Polarity, acidity and viscosity may all act to influence the relative accessibility and stability of the charge transfer (CT) state and the kinetics of its population. Molecular aspects of solvation can be studied by the investigation of isolated gas phase “ solute–solvent” clusters, which are formed in a supersonic jet expansion. Among the issues concerning the excited state CT process are the critical number of solvent partners of a defined polarity, the contributions of the local and long-range stabilizing forces, and details of the solute–solvent interactions, e.g., steric effects, the solvation site(s), the solvent orientation and the role of hydrogen bonding.
Experiments with clusters in a supersonic jet are advantageous for studying the effects of microsolvation of a chromophore on its emission spectrum, especially if the composition and the structure of the solute–solvent clusters can be determined. In this article the emission behaviour of three aminobenzonitriles complexed with various solvent molecules (such as acetonitrile, methanol, tetrahydrofuran, cyclohexane and n-hexane) in a supersonic jet is reported. The chromophores studied are 4-dimethylaminobenzonitrile (DMABN), 4-aminobenzonitrile (ABN) and 1-ethyl-2,3-dihydro-indole-5-carbonitrile, EIN (Fig. 1). In contrast to DMABN, both ABN and EIN do not exhibit dual fluorescence in solution. This has been taken as major evidence for the TICT mechanism.3,4 The cluster size distributions are determined by time-of-flight mass spectrometry (TOF-MS) after resonant two-photon ionization (R2PI). This allows for a better characterization of the cluster sizes probed in the emission experiments than correlations with expansion parameters such as the nozzle temperature, the partial pressure of the solvent and stagnation pressure used in previous studies.7–16 Emission spectra of DMABN7–15,17 and ABN10,16 solute–solvent complexes have been already studied by various groups. Mass-resolved experiments on DMABN·(solvent)n clusters, henceforth abbreviated as 1:n complexes, were also reported by Bernstein et al.17,18 The emission spectrum of the bare EIN molecule in a supersonic jet has been reported before by our group.19
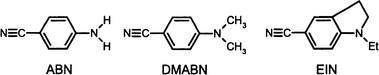 | ||
| Fig. 1 Molecules studied and their abbreviations used in the text. | ||
2. Experimental
The experimental setup for the supersonic jet experiments has been described in detail in ref. 20. Briefly, the UV excitation is provided by the pulsed, frequency-doubled output of a Nd:YAG pumped optical parametric oscillator OPO (Continuum, Sunlite) running at a repetition rate of 10 Hz. With helium as the carrier gas, the chromophore and solvent molecules are expanded by a pulsed nozzle into two sequentially aligned vacuum chambers, which are connected![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) ia a skimmer. The low-pressure solvent vapour is mixed with the carrier gas before introducing it into the nozzle. Its amount is controlled with needle valves regulating the flow conditions and by the temperature of the solvent reservoir. In the first vacuum chamber (expansion chamber) the clusters are excited by
the laser radiation in order to observe the dispersed emission spectra. Fluorescence is collected at a right angle to both the jet and the laser beam and detected with a CCD-camera coupled to an image intensifier (LaVision, FL-III). Before detection the emission is dispersed by a spectrograph (Chromex 250 IS) with a 300 grooves mm−1 grating used in first
order. The spectral range covered is ∽200 nm and the spectra were accumulated for 2–3 min (spectral resolution ∽0.3 nm). In the second chamber, ionization chamber, the clusters
are ionized by one-color resonant two-photon ionization ia a resonant
transition into the S1 vibronic manifold and a non-resonant second step into the ionization continuum. Subsequently they are mass analyzed in a home-built reflectron time-of-flight mass spectrometer. A variable time delay between laser pulse and valve opening pulse is used to ensure that the
same portion of the inhomogeneous gas pulse is probed for the
dispersed emission spectrum in the first chamber and for
the cluster size analysis in the second chamber (correction of the flight time of the molecules between the two
chambers). The intensities of laser excitation for mass and emission spectra were kept equal (∽106 W cm−2).
ia a skimmer. The low-pressure solvent vapour is mixed with the carrier gas before introducing it into the nozzle. Its amount is controlled with needle valves regulating the flow conditions and by the temperature of the solvent reservoir. In the first vacuum chamber (expansion chamber) the clusters are excited by
the laser radiation in order to observe the dispersed emission spectra. Fluorescence is collected at a right angle to both the jet and the laser beam and detected with a CCD-camera coupled to an image intensifier (LaVision, FL-III). Before detection the emission is dispersed by a spectrograph (Chromex 250 IS) with a 300 grooves mm−1 grating used in first
order. The spectral range covered is ∽200 nm and the spectra were accumulated for 2–3 min (spectral resolution ∽0.3 nm). In the second chamber, ionization chamber, the clusters
are ionized by one-color resonant two-photon ionization ia a resonant
transition into the S1 vibronic manifold and a non-resonant second step into the ionization continuum. Subsequently they are mass analyzed in a home-built reflectron time-of-flight mass spectrometer. A variable time delay between laser pulse and valve opening pulse is used to ensure that the
same portion of the inhomogeneous gas pulse is probed for the
dispersed emission spectrum in the first chamber and for
the cluster size analysis in the second chamber (correction of the flight time of the molecules between the two
chambers). The intensities of laser excitation for mass and emission spectra were kept equal (∽106 W cm−2).
ABN and DMABN were obtained from Fluka, while EIN was synthesized according to the procedure described in ref. 21.
3. Results
The cluster size distribution in the supersonic jet was varied by adjusting the nozzle temperature, the electric driving pulse for the nozzle, the time delay between nozzle and laser pulse, and the partial pressure of the solvent in the He carrier gas. Each time, TOF-MS was used to determine the cluster sizes that were excited in the dispersed emission experiment. Because the clusters are ionized by a one-color-R2PI scheme, it is expected that cluster fragmentation can not be neglected. This is due to the excess energy in the photoionizing step, which may, in spite of the propensity rule, not be completely carried away by the photoelectron. Another reason is the low binding energy in a heterogeneous van der Waals (vdW) or hydrogen-bonded complex consisting of a chromophore and solvent molecules. Therefore, the mass spectrum is not fully representative for the cluster size distribution in the jet. Within this limitation two different (chromophore)m–(solvent)n cluster size distributions were studied: (i) complexes containing one chromophore molecule (m = 1) and a small number of solvent molecules (n = 1–4), and (ii) complexes of one chromophore and a large number of solvent molecules (n = 1–12). Illustrative mass spectra for these two different cluster size distributions are shown in Fig. 2 for the EIN–acetonitrile complexes. For ABN and DMABN similar cluster size distributions were investigated. The low nozzle temperatures ensured that homogeneous dimer formation (self-complexation) was negligible in the jet. This was confirmed by the complete absence of the homogeneous dimer signal in the mass spectrum (except for DMABN). Photoexcitation of homogeneous clusters (self-complexes) leads to the formation of excimers, which display dual fluorescence.20 Thus, to avoid interference of excimer emission with the emission from the heterogeneous clusters, the formation of homogeneous dimers should be suppressed. Excimer formation in a cluster is largely facilitated in comparison to the liquid phase because of the lower temperature and the close proximity of the second chromophore. | ||
| Fig. 2 R2PI TOF mass spectra of a EIN + acetonitrile expansion for two different expansion conditions: (a) stagnation pressure 4 bar He, nozzle temperature 80°C, (b) stagnation pressure 5 bar He and larger flow of solvent vapour, nozzle temperature 80°C. Excitation wavelength for both spectra is 325.60 nm (30713 cm−1) (0,0 − 40 cm−1 transition of the bare monomer). | ||
The dispersed emission spectra of the jet-cooled chromophores ABN, EIN and DMABN complexed with a varying number of solvent molecules are shown in Figs. 3 and 4. The respective excitation wavelengths were slightly red-shifted relative to the 0,0 transitions of the bare monomers as indicated in the figure captions (blue-shifted for DMABN, see below). In general no characteristic features in the R2PI spectra for the larger clusters could be found. Because for the small clusters (1:1) no changes occur, those excitation wavelengths have been chosen to excite a variety of the larger clusters simultaneously and non-resonantly.
 | ||
| Fig. 3 (a) Dispersed fluorescence spectra of ABN complexed with a small number (n = 1–4) of solvent molecules such as THF and acetonitrile. Additionally, the emission spectrum of the bare monomer is shown. Excitation wavelength is 300 nm (33333 cm−1), i.e. 0,0 − 160 cm−1 transition of the bare monomer. Stagnation pressure 4 bar He, nozzle temperature 85°C. (b) Dispersed fluorescence spectra of EIN complexed with a small number (n = 1–4) of solvent molecules such as cyclohexane, THF and acetonitrile. Additionally, the emission spectrum of the bare monomer is shown. Excitation wavelength is 325.60 nm (30713 cm−1), i.e. 0,0 − 40 cm−1 transition of the bare monomer. Stagnation pressure 4 bar He, nozzle temperature 80°C. | ||
 | ||
| Fig. 4 Dispersed fluorescence spectra of a supersonic expansion with DMABN and acetonitrile and the corresponding mass spectra, which show the clusters size distribution for the emission experiment. Excitation wavelength is 306.24 nm (32654 cm−1) (the vibronic transition of the 1:1 complex), except for the emission spectrum at the top where an excitation wavelength of 308.70 nm (32394 cm−1) was used (the vibronic transition of the bare monomer). Stagnation pressure 1.5–2.5 bar He, nozzle temperature 80°C. Left: the dispersed fluorescence spectra of DMABN resulting from the formation of different self complexes and heterogeneous clusters. The fluorescence can be illustrated by four spectra corresponding to: (a) monomer, (b) monomer, 1:n (n⩽5) complexes and dimer, and (c and d) monomer, 1:n (n⩽8) complexes, dimer and its small clusters. Right: the corresponding mass spectra are shown. | ||
In agreement with previous studies7–10,12,14–16 it is found that solute–solvent 1:1 complexes do not show any significant change and shift of the emission spectra of the bare chromophores which could be attributed to emission from the CT state. However, complexation with several solvent molecules (n>1) changes this behaviour. The emission spectra of ABN complexed with a small number (n = 1–4) of solvent molecules such as acetonitrile and tetrahydrofuran (THF) are shown in Fig. 3a. Fig. 3b shows the emission spectra of EIN complexed with acetonitrile, THF or cyclohexane (the corresponding mass spectra for the employed expansion conditions are similar to that shown in Fig. 2a). For both molecules the red-shift of the emission maximum with respect to the uncom plexed chromophore increases with increasing polarity of the solvent molecules. The shift is largest (∽2200 cm−1) for the complexes with the highly polar acetonitrile and smallest (∽200 cm−1) for the nonpolar cyclohexane. The spectral positions of the emission maxima of the jet-cooled solute–solvent complexes are summarized in Table 1, together with the solvent induced shifts. For comparison, also the fluorescence maxima in solution are given. It is noteworthy that the emission maxima of the complexed chromophores in the jet resemble closely those in the respective solutions. Particularly, no second, anomalous and red-shifted emission band is found under these expansion conditions (i.e. when only monomers (m = 1) and complexes with n = 1–4 solvent molecules are formed).
![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 5 for DMABN) and in the corresponding solution phase. For the jet spectra the red-shift in cm−1 with respect to the monomer emission is also indicated. Excitation wavelengths are given in the figure captions. Corrected emission spectra in solution were measured using a Perkin-Elmer 650-60 spectrofluorimeter
5 for DMABN) and in the corresponding solution phase. For the jet spectra the red-shift in cm−1 with respect to the monomer emission is also indicated. Excitation wavelengths are given in the figure captions. Corrected emission spectra in solution were measured using a Perkin-Elmer 650-60 spectrofluorimeter
No significant changes in the emission spectrum are observed when one increases the size of the “solvent shell” (i.e. from n⩽4 to n⩽12). Occasionally, a very small additional shift (∽5 nm) and a slight broadening of the emission band have been found under increased microsolvation.
DMABN–solvent clusters show a different behaviour. According to refs. 17 and 18 DMABN forms two structural isomers of 1:1 complexes with acetonitrile. One isomer exhibits a structured excitation spectrum, similar to that of the bare molecule, but blue-shifted by about 252 cm−1, while the other exhibits a more congested and red-shifted excitation spectrum. The vibrational structure in the former region has been also observed in our experiments. The influence on the fluorescence spectrum when DMABN is complexed with acetonitrile is shown in Figs. 4 and 5. The monomer emission is centred at about 330 nm. At expansion conditions favouring the
formation of monomers and 1:1 clusters between DMABN and acetonitrile and upon excitation at the most prominent vibrational band9,17,18 of the “blue” 1:1 cluster (i.e. at 306.24 nm), the emission spectrum shows two maxima centred
around 325 nm and 345 nm. The latter maximum is assigned to the 1:1 complex.17 The formation of a small amount of dimers (2:
0) and 1:n (n⩽4) solute–solvent complexes in
the jet (as it is observed in the corresponding TOF mass spectrum) changes the position and shape of the luminescence spectrum with respect to that of the monomer and 1:1 complexes only slightly. The behaviour is very similar upon excitation at the 0,0 transition of the monomer (about 310.0 nm)
and at the positions of the respective vibrational bands as well as in the region of either the “blue” or the “red” 1:1 cluster (around 318 nm).17,18 In contrast, the formation of 1:
n clusters with n5 seems to give rise to a strongly red-shifted and clearly separated second emission band with a maximum around 425 nm. From the mass spectrum, however, one knows that the emission spectra of the dimers (with maximum at about 370–390 nm)20 and the 2:n complexes could be additionally superimposed in the latter band. In order to assign the long wavelength emission to clusters of known stoichiometry the
relative increase of the integrated intensity of this band has been correlated with the relative concentration of various
mass-analyzed ionic clusters of 1:
n and 2:n stoichiometry
(Fig. 5). The relative concentration is expressed by the ratio of the intensity of the mass signal of the particular ion cluster and the total intensity of all signals in the mass spectrum. It
should be pointed out that this procedure has been chosen due to the fact that the excitation spectrum of this fluorescence band is diffuse and does not
exhibit any pattern of characteristic features.17,18Fig. 5 clearly indicates that the emission band at 425 nm can be assigned to the CT state of the 1:n
complexes with n5. On the other hand the CT emission
intensity shows nearly no dependence on the relative concentration of the DMABN dimer and its complexes. The corresponding spectra of the DMABN–methanol and DMABN–THF clusters are qualitatively very similar, but show different shape and spectral position of the long wavelength band (see Table 1). Dual luminescence is observed also for the DMABN dimer complexed with n-hexane but not for
the 1:
n complexes with n-hexane.
 | ||
| Fig. 5 Correlation between the relative increase of the integrated intensity of the long wavelength emission band of the DMABN–acetonitrile clusters (see Fig. 4) and the relative concentration of selected cluster sizes, with stoichiometries of 1:1, 1:4, 1:5, 1:6, 2:0 and 2:3. The behaviour of the 1:2 and 1:3 complexes is similar to that of the complexes with 1: 4 stoichiometry, and the correlation for the 2:1, 2:2 and 2:4 clusters is nearly the same as that for the 2: 3 clusters. The relative concentration is expressed by the ratio of the intensity of the mass signal of the particular ion cluster and the total intensity of all ion signals in the mass spectrum. Both the intensity of the red-shifted emission band and the concentration of the ions is related to the spectrum where this emission band starts to appear (Fig. 4b). | ||
4. Discussion
In the previous section the emission spectra of mixed clusters consisting of aminobenzonitriles and various solvent molecules were presented. The questions addressed in this section pertain to the nature of the excited states of these donor–acceptor species and the interactions responsible for the different spectroscopy and photophysics under microsolvated conditions. A comparison of the spectra of small clusters with those in solution allows one to elucidate the approximate minimum cluster size that exhibits the same behaviour as the chromophore in solution.The emission spectra of jet-cooled ABN–solvent and EIN–solvent complexes show a solvent dependent red-shift with respect to the emission of the bare monomer. The magnitude of the red-shift increases with increasing polarity of the solvent molecules and resembles closely the red-shift in solution with the respective solvent. By the combination of emission spectroscopy and TOF-MS the number of solvent molecules sufficient to induce an emission behaviour similar to that observed in solution could be determined to 2–4 solvent molecules.
In agreement with Phillips et al.14 dual fluorescence for the
jet-cooled DMABN is observed for the bare and the solvated dimer (it has been shown previously that this dual fluorescence of the dimer may be rationalized by the formation of excimers20).
However, in contrast to their observations, long wavelength fluorescence is also found for the monomer complexed with five or more acetonitrile molecules (or other polar solvent molecules such as methanol and THF). This follows from
the analysis of the mass spectra, a technique which was not available in the experiments by Phillips et al. Furthermore, the red-shift of the bare excimer emission of DMABN is significantly smaller than that for the DMABN–(solvent)n (n5) complexes. These findings on microsolvated monomers agree nicely with the reported value of 5 solvent molecules in the
case of the DMABN–acetonitrile clusters,17 which are sufficient to observe an R2PI spectrum similar to the absorption spectrum of DMABN dissolved in acetonitrile.22
Moreover, in accordance with previous studies,7–9,12,14,15 it is confirmed that one polar solvent molecule is not sufficient to observe the long wavelength emission band that can be assigned to CT fluorescence. It is noteworthy that this result is different from the behaviour of microhydrated dimethylaminobenzoic acid methylester (DMABME), as reported by Dedonder–Lardeux et al.,15
where complexation with two (or even one) water molecules results in the appearance of the CT emission band.
While for the microsolvated clusters of ABN and EIN only a single red-shifted fluorescence band appeared, those of DMABN showed two bands for larger clusters. This striking difference in the photophysics deserves careful consideration. To rationalize these findings, a qualitative potential energy model is presented in Fig. 6 both for molecules with and without a CT transition.
 | ||
| Fig. 6 Potential energy surfaces of the ground and excited state relevant to the different emission behaviour of DMABN (a) and ABN and EIN (b). See text for explanation. | ||
Excitation of a vdW cluster leads to the primary excited state (vdW)* from which the excited state of the photoinduced reaction product can be populated. The equilibrium conformation of the vdW cluster and the shape of the potential energy curves in the ground and excited state are usually dif ferent due to a different binding energy in the excited state with respect to the ground state. This could result from the increase of the dipole moment of the solute molecule and/or from exciton or CT interactions. The actual dynamics of a particular, electronically excited vdW complex depends on the electronic structure of the chromophore (e.g., on the energy gap between the interacting electronic states) and on the intracluster interactions (which may strongly depend on the electron donor–acceptor properties of the solute and the equilibrium geometry of the solute–solvent clusters). The relatively small red-shift of the emission of ABN and EIN solute–solvent clusters with polar partners as compared to that of the monomers and clusters with nonpolar molecules (Fig. 3) is most probably caused by the change of the potential energy surface of the vdW cluster together with intramolecular vibrational redistribution (IVR). By the latter various low-frequency vibronic states in the S1 state would be populated (Fig. 6b).
The striking appearance of dual luminescence in DMABN
clusters with polar molecules must be of different origin. Following
the arguments given by Bixon et al.23,24 Phillips et
al.,14 Wersink and Wallace,25 and Yip and Levy,26 the origin
of a new red-shifted emission band can be due to: (i) the formation of an exciplex (1:n complexes, n5) and (ii) a solvated excimer (2:
n complexes), and/or (iii) the transition into an intramolecular CT state of the solute, which is induced by its polar
partners (Fig. 6a). Most likely the latter mechanism is responsible for the long wavelength luminescence of jet-cooled 1:n
DMABN–solvent complexes. The easier accessibility of the CT
state in DMABN is most probably related to the lower ionization potential of DMABN as compared to ABN, with
this energy difference amounting to at least 0.6 eV.27,28 This
value may be even larger in the case of the formation of a TICT
state due to the decoupling of the amino group. Thus, relatively strong interactions between the primary excited state (S1) and the CT state (S2) due to their small energy gap are expected for DMABN (see eqn. (1) in ref. 20 and refs. 4 and 29). The excimer origin of the long wavelength emission band
is excluded by its appearance in solvated monomers of the chromophore. The exclusion of the exciplex origin of this emission is supported by the relatively low electron affinity and high ionization potential of the solvent molecules, although it should be admitted that larger solvent clusters show
a decreased ionization potential30 and an increased electron
affinity31 in comparison to a bare solvent molecule. Moreover, if such DMABN–solvent exciplexes correspond to the planar conformation, bridged compounds such as EIN should show similar solute–solvent exciplexes with the corresponding
red-shifted emission.
An additional reason for the CT population in DMABN might be related to IVR. The role of IVR on CT processes has been studied by Itoh and Kajimoto.32 They found that an enhanced IVR rate leads to an increase in the CT rate. The enhancement of IVR was attributed to the presence of methyl groups, which act as the promoting internal rotors. A similar effect could be of importance here. The increasing extent of IVR in aminobenzonitriles can be seen in the emission spectra of the monomers. In a series of aminobenzonitriles with an increasing number of methyl groups (e.g., ABN, DMABN, 4-dimethylamino-3-methylbenzonitrile) an increase of congestion and bandwidths in the jet emission spectra is observed (see e.g., ref. 20).
The accessibility of the CT state in the DMABN–solvent clusters leads to the question of the origin of dual fluorescence for this molecule. The difference in the emission spectra between DMABN and EIN is tempting to attribute the dual fluorescence to a twisting motion of the dimethylamino group in terms of the TICT model. However, it should be pointed out that EIN, used as a non-TICT model compound in discussions of the TICT mechanism, due to its blocked twist of the amino group, shows, relative to DMABN, a large decrease in energy of its S1 state. For example, the origin band of EIN is red-shifted by about 1500 cm−1 with respect to that of DMABN and strong spectral congestion sets in from the very beginning of the spectrum.19,20 The exact energy of the S2 state is unknown. Thus, although the relative sequence of excited states is the same in EIN and DMABN as judged from fluorescence polarization experiments,33,34 the quantitative correspondence of the electronically excited states in these molecules is very uncertain. Therefore other reasons, besides the restricted conformational flexibility, could contribute to the different luminescent properties.
Another complication in any mechanistic discussion arises from the complexity of the reaction coordinate. The reaction coordinate of an intramolecular CT reaction in the chromophore embedded in a stoichiometric cluster should be complex, i.e. it should depend presumably on the intermolecular distance and the mutual orientation of the partners and possibly on intramolecular torsional and inversion motions in the donor–acceptor solute molecule.
Although the present results clearly indicate a reaction mechanism which is intramolecular in the solute, final conclusions regarding the details of the mechanism cannot be drawn yet. Discrimination between the various reaction mechanisms proposed in the literature, such as the TICT-,2,3 the solvent-induced pseudo-Jahn–Teller coupling-,5 and even the exciplex mechanism,6 is not possible at the present stage of cluster studies. However, the differences in the photophysics of EIN and DMABN could be indicative of the important role of the twisting coordinate.
A surprising result in this study is the low number of solvent molecules that are sufficient to induce a liquid phase-like emission. However, the mechanisms responsible for the red-shifted emission in a supersonic jet and in the solution are different. Under the collision free conditions, a fluorescence transition from a CT state with excited vibrational degrees of freedom should take place under conservation of the vibrational energy. The difference between the conformation of the CT state with respect to that of the vdW cluster in the ground state generates a broad, red-shifted emission (Fig. 6a). The effect is similar to that in solutions. In solution, however, the solvent effects on fluorescence can be rationalized in terms of the Onsager theory of continuum dielectric polarization.35 Here the solvatochromic shift of the fluorescence spectral position is largely due to the reorientation of the solvent molecules in combination with a highly efficient energy transfer to the bulk solvent.
A critical evaluation of the findings of this work needs some further remarks on the possible energy dissipation processes in clusters and in the solution phase. As already mentioned, the determination of the cluster size distribution in the beam by R2PI-TOF-MS is limited by the intermolecular fragmentation of the vdW clusters in the ionic and possibly also in the S1 state. It should be pointed out that clusters between aromatic chromophores and water, methanol, and other polar molecules usually dissociate upon one-color-R2PI by losing one solvent molecule.36 This fragmentation is mostly due to an excess energy left in the ion after a non-resonant ionization transition and/or by a rearrangement of the ionized cluster due to the charge-dipole interaction. The absence of the emission of the bare monomer for the ABN/EIN–solvent clusters suggests that no (or only very little) fragmentation down to the bare monomer occurs in the S1 state prior to the fluorescence transition. However, if an exothermic reaction (e.g., a CT process) takes place (Fig. 6a), in addition to the fragmentation after ionization, an unknown amount of fragmentation could happen in the electronically excited state (S1). The system transforms a certain amount of electronic energy into vibrational energy which could be imparted by IVR to the solvent molecules, followed by a fast predissociation of one or even several of them. Energy dissipation by fragmentation could be inefficient, if the number of coupled vibrational degrees of freedom of the complex increases, as typical in larger clusters. Thus, if such a complex is unable to further fragment, IVR and internal conversion (IC) could be dominant dissipation channels favouring a transition into a CT state.
5. Concluding remarks
The small number of solvent molecules sufficient for the observation of dual fluorescence points to a very active role of the solvent in the photophysics of the compounds studied. Although it has been confirmed that the emission spectra of the 1:1 solute–solvent complexes do not show any significant changes with respect to those of the bare chromophores, complexation with several molecules results in a red-shifted emission. 2–4 solvation partners induce the emission spectra of ABN and EIN similar to those observed in the respective solution. Five or more acetonitrile (and methanol or THF) molecules complexed with DMABN lead to the long wavelength emission which is attributed to the formation of the CT state. The different luminescence properties of DMABN with respect to ABN/EIN solute–solvent complexes are most probably related to the lower ionization potential of the former molecule and/or more efficient IVR and IC processes in the respective clusters.In the present experiment in spite of a mass spectrometric analysis of the clusters in the molecular beam by R2PI, the role of fragmentation in the energy dissipation of the electronically excited cluster is not yet well defined. Experiments allowing for a better discrimination between the contributions of a specific cluster size and the intracluster CT process could give further information on the nature of the CT mechanism. Similarly a more detailed insight into the conditions of the electronic states, which are required for a photoinduced, solvent mediated charge transfer is necessary. In this respect new methodological developments are needed. One of the methods which may allow one to characterize the size and structure of the vdW complexes undergoing excited-state electron transfer is double resonance IR/R2PI laser depletion spectroscopy (e.g., ref. 30). This method is presently being developed in this lab.
Acknowledgements
We acknowledge the funding of this work by the Deutsche Forschungsgemeinschaft (SFB 337) and by the “Fonds der Chemischen Industrie”. The authors thank Armin Gerlach, Andrea Schmidt and Ulrich Verleger for experimental assistance and Barbara Bliß for synthesizing EIN.References
-
E. Lippert, W. Lüder and H. Boos, Ad. Mol. Spectrosc., Proc. Int. Meet. 4th, A. Mangini, Pergamon, Oxford, 1962, p. 443. Search PubMed.
- K. Rotkiewicz, K. H. Grellmann and Z. R. Grabowski, Chem. Phys. Lett., 1973, 19, 315 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz, A. Siemiarczuk, D. J. Cowley and W. Baumann, Nou. J. Chim., 1979, 3, 443 Search PubMed.
- W. Rettig, Angew. Chem. Int. Ed. Engl., 1986, 98, 969 CAS.
- K. A. Zachariasse, M. Grobys, Th. von der Haar, A. Hebecker, Yu. V. Il'ichev, Y.-B. Jiang and O. Morawski, J. Photochem. Photobiol. A, 1996, 102, 59 Search PubMed.
- R. J. Visser and C. A. G. O. Varma, J. Chem. Soc., Faraday Trans 2, 1980, 76, 453 RSC.
- E. M. Gibson, A. C. Jones and D. Phillips, Chem. Phys. Lett., 1987, 136, 454 CrossRef CAS.
- T. Kobayashi, M. Futakami and O. Kajimoto, Chem. Phys. Lett., 1986, 130, 63 CrossRef CAS.
- L. W. Peng, M. Dantus, A. H. Zewail, K. Kemnitz, J. M. Hicks and K. B. Eisenthal, J. Phys. Chem., 1987, 91, 6162 CrossRef CAS.
- E. M. Gibson, A. C. Jones, A. G. Taylor, W. J. Bouwmann, D. Phillips and J. Sandell, J. Phys. Chem., 1988, 92, 5449 CrossRef CAS.
- J. August, T. F. Palmer, J. P. Simons, C. Jouvet and W. Rettig, Chem. Phys. Lett., 1988, 145, 273 CrossRef CAS.
- J. Herbich, F. Pérez-Salgado, R. P. H. Rettschnick, Z. R. Grabowski and H. Wójtowicz, J. Phys. Chem., 1991, 95, 3491 CrossRef CAS.
- B. D. Howells, J. McCombie, T. F. Palmer, J. P. Simons and A. Walters, J. Chem. Soc., Faraday Trans., 1992, 88, 2587 RSC.
- R. Howell, D. Phillips, H. Petek and K. Yoshihara, Chem. Phys., 1994, 188, 303 CrossRef CAS.
- C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard, D. Solgadi, J. McCombie, B. D. Howells, T. F. Palmer, A. Subaric-Leitis, C. Monte, W. Rettig and P. Zimmermann, Chem. Phys., 1995, 191, 271 CrossRef CAS.
- E. M. Gibson, A. C. Jones and D. Phillips, Chem. Phys. Lett., 1988, 146, 270 CrossRef CAS.
- S. Quan-Yuan and E. R. Bernstein, J. Chem. Phys., 1992, 97, 60 CrossRef.
- J. A. Warren, E. R. Bernstein and J. I. Seeman, J. Chem. Phys., 1988, 88, 871 CrossRef CAS.
- U. Lommatzsch and B. Brutschy, Chem. Phys., 1998, 234, 35 CrossRef CAS.
- U. Lommatzsch, C. Lahmann, A. Gerlach and B. Brutschy, J. Phys. Chem. A, 1998, 102, 6421 CrossRef CAS.
- K. Rotkiewicz, Z. R. Grabowski, A. Krówczynski and W. Kühnle, J. Lumin., 1976, 12/13, 877 CrossRef.
- C. Cazeau-Dubroca, S. Ait Lyazidi, P. Cambou, A. Peirigua, Ph. Cazeau and M. Pesquer, J. Phys. Chem., 1989, 93, 2347 CrossRef CAS.
- M. Bixon and J. Jortner, J. Phys. Chem., 1993, 97, 13061 CrossRef CAS.
- J. Jortner, M. Bixon, H. Heitele and M. E. Michel-Beyerle, Chem. Phys. Lett., 1992, 197, 131 CrossRef CAS.
- R. A. Wersink and S. C. Wallace, J. Phys. Chem., 1994, 98, 10710 CrossRef.
- W. T. Yip and D. H. Levy, J. Phys. Chem., 1996, 100, 11359 CrossRef.
- A. Modelli and G. Distefano, Z. Naturforsch. A, 1981, 36, 1344 Search PubMed.
- A. Buchs, Hel. Chim. Acta, 1970, 53, 2026 Search PubMed.
- A. Weller, Z. Phys. Chem. NF, 1982, 133, 93 Search PubMed.
- B. Brutschy, Chem. Re., 1992, 92, 1567 Search PubMed.
- Ch. Desfrancois, H. Abdoul-Carime and J.-P. Schermann, Int. J. Mod. Phys., 1996, 10, 1339 Search PubMed.
- M. Itoh and O. Kajimoto, in Dynamics of excited molecules, ed. K. Kuchitsu, Elsevier, Amsterdam, 1994, p. 333, and references cited therein. Search PubMed.
- W. Rettig, G. Wermuth and E. Lippert, Ber. Bunsen-Ges. Phys. Chem., 1979, 83, 692 Search PubMed.
- G. Wermuth and W. Rettig, J. Phys. Chem., 1984, 88, 2729 CrossRef CAS.
- L. Onsager, J. Am. Chem. Soc., 1936, 58, 1486 CrossRef CAS.
- H.-D. Barth, K. Buchhold, S. Djafari, B. Reimann, U. Lommatzsch and B. Brutschy, Chem. Phys., 1998, 239, 49 CrossRef CAS.
Footnote |
| † Present address: Department of Chemistry, Stanford University, Stanford, USA. |
| This journal is © the Owner Societies 2001 |