Energy barrier to TTC–TTT isomerisation for the merocyanine of a photochromic spiropyran
Jonathan Hobley†* and Vincenzo Malatesta
Great Lakes Chemical Italia, Via Maritano 26, 20097, San Donato Milanese, Italy
First published on UnassignedUnassigned22nd December 1999
Abstract
The 1H NMR signal of a proton of a spiropyran photomerocyanine was resolved at low temperature into two signals attributed to the two isomers, TTT and TTC. The equilibrium distribution and free energy difference between the TTT and TTC isomers were calculated to be 0.15 and 4.6±0.7 kJ mol−1, respectively. By 1H NMR lineshape analysis at various temperatures, an activation barrier for the TTC→TTT isomerisation of 43.6±3 kJ mol−1 was calculated.
Introduction
Photochromism of spiropyrans to give the corresponding photo-merocyanines is based on the reaction shown in Scheme 1. | ||
| Scheme 1 | ||
For many years, there has been speculation that isomeric distributions of spiropyran and spirooxazine merocyanines exist in equilibrium in different solvents.1–8 Such arguments have been widely used to explain the results obtained from picosecond and nanosecond transient spectroscopy1–4,6–8 and from steady-state spectroscopy.5 In the transient experiments spectral evolution as a function of time has often been explained in terms of equilibration of merocyanine isomers to form the thermodynamically favoured distribution of isomers in the particular solvent being studied, but actual equilibrium distributions have never been established.
Work carried out in these laboratories9 has shown that isomeric distributions of spiropyran merocyanines do exist in different solvents with only the TTC and TTT isomers implicated and the TTC isomer dominating. Further, we have observed temperature dependent 1H NMR line broadening in chloroform solutions of the spiropyran 6,8-dinitro-BIPS that is indicative of rotation about the bond on the methine bridge at a rate in the intermediate region on the 400 MHz 1H NMR time-scale. Unfortunately, in those studies we were unable to resolve the broadening peaks into their composite signals owing to the freezing-point of this solvent, a factor which limited the conclusions of that work. In the present study, we worked with 6-nitro-8-bromo-BIPS, which in methylene chloride solution demonstrates the same line broadening characteristics as 6,8-dinitro-BIPS does in chloroform, except that the broadening occurs at higher temperature. For this reason it was possible, within the liquid range of this solvent, to resolve the two separate resonances for the H-5B proton for the TTC and TTT isomeric forms.
Experimental
6-Nitro-8-bromo-BIPS was prepared using standard preparative methods.10 Its purity was established by 1H NMR spectroscopy.6-Nitro-8-bromo-BIPS crystallises from methanol as the merocyanine form of the molecule and dissolves in methylene chloride to form a merocyanine solution. Over a period of hours this solution equilibrates back to the spiropyran form even in dark conditions, but the merocyanine form can be trapped long enough for one-dimensional spectra to be obtained by cooling the solution to 243 K as soon as it is dissolved. 1H NMR spectroscopy can then be carried out on this solution at temperatures below 243 K. However, conversion to the closed form still occurs even at these low temperatures, albeit slowly.
1H NMR experiments were carried out on a Varian VXR 400 MHz spectrometer in 99.6% d2-methylene chloride solution (Cortec). Spectra were collected over a range of temperatures from 243 to 183 K.
NOE difference experiments were carried out on C-3 protonated and deuterated forms of 6-nitro-8-bromo-BIPS in 99.96% d6-DMSO (Cortec) using a Bruker NR 200 AF 200 MHz spectrometer. Mixing times between 1 and 3 s were used. The program G-NMR available from Cherwell Scientific was used to simulate the results obtained at the different temperatures and extract kinetic information as a function of temperature.
Results and discussion
H NMR peak positions and assignments and comments in methylene chloride are given in Table 1. The peaks were assigned on the basis of coupling, integrals and multiplicity and a knowledge of the data collected for 6,8-dinitro-BIPS and reported previously.9 The only ambiguity in these assignments was between the methine protons, H-3B and H-4B, on the bridging region of the merocyanine, but it was found that the 1H NMR signal at 8.88 ppm was relatively diminished, as observed over a period of hours, on contacting this solution in methylene chloride with deuterium oxide. The closed form spectrum that evolved due to thermal equilibration of the spiropyran and merocyanine forms also had a diminished H-3A resonance integral. In deuterated methanol in which the merocyanine form dominates, deuterated 6-nitro-8-bromo-BIPS was formed by exchange of the alcohol deuteron with H-3B. 3-Deuteron-6-nitro-8-bromo-BIPS was then recrystallised from this solution and dissolved in methylene chloride. In this case the peak at 8.88 ppm was completely absent. As has been reported previously, H-3B on the methine bridge of spiropyran merocyanines is labile and can be exchanged fairly easily with deuterated water added to the solvent.9,11 These observations and findings allowed us to assign unambiguously the H-3B resonance as that at 8.88 ppm.| gem-Me | N-Me | 4′B | 5′B | 6′B | 7′B | 3B | 4B | 5B | 7B |
|---|---|---|---|---|---|---|---|---|---|
| a Note that where peaks are exchange broadened, the centre of the peak has been approximated. S=singlet; O=overlapping other resonances; D=doublet. | |||||||||
| 1.75 | 3.91 | 7.52 | 7.45 | 7.45 | 7.37 | 8.88 | 8.04 | 8.25 | 8.36 |
| S | S | O | O | O | D | Broad | Broad | Broad | D |
| T/K | 243 | 233 | 213 | 193 | 183 |
|---|---|---|---|---|---|
| k/s−1 | 0.102±0.015 | 0.526±0.079 | 3.89±0.039 | 52.2±5.22 | 92.0±9.2 |
From NOE difference experiments on 6-nitro-8-bromo-BIPS in DMSO solution, the predominant merocyanine isomer was found to be the TTC isomer since a stronger NOE was observed between the H-5B and H-4B resonances than between the H-5B and H-3B resonances. Strong coupling between the H-4B and H-3B resonances in DMSO makes it impossible to assign clearly the presence of the TTT isomer in this instance. This is because through-bond transfer of the NOE12 undoubtedly occurs in this situation and an NOE would inevitably be seen between H-5B and H-3B owing to H-3B's strong coupling to H-4B. From NOE difference experiments on the C-3-deuterated form of 6-nitro-8-bromo-BIPS in DMSO, for which H-4B is no longer strongly coupled, it was possible to conclude that isomers cis about the α-bond were not present in DMSO solution, since no NOE was detected between the N-methyl protons and H-4B. It was also possible to confirm the presence of TTC owing to an NOE between H-4B and H-5B. From a knowledge of the behaviour of 6,8-dinitro-BIPS9 and the fact that for this compound in all solvents investigated the TTC isomer dominated, we feel that it is reasonable to assume that the TTC isomer is also dominant in methylene chloride. Unfortunately, the actual NOE and NOESY experiments in this solvent are impractical owing to exchange broadening of the relevant resonances, low merocyanine solubility and the requirement for low temperatures to freeze in the merocyanine form, albeit temporarily. All of these factors make one- and two-dimensional NOE experiments impractical. Even for the one-dimensional spectra obtained here some 500–600 transients were required for a single spectrum.
A second isomer was also identified in methylene chloride solution from the temperature dependence of the 1H-NMR spectra (see below). A knowledge that in DMSO solution isomers cis about the α-bond are not significant and by inference from observing those resonances which are affected by the temperature variation it is logical to assume that this second isomer is TTT if the dominant isomer is TTC. We also reasonably expect the TTT isomer to be in equilibrium with the TTC isomer from a knowledge of the behaviour of 6,8-dinitro-BIPS in all solvents investigated and the similarity of the entire exchange broadening phenomena for these two compounds.9
The temperature dependence of the merocyanine form of 6-nitro-8-bromo-BIPS is shown in Fig. 1. As can be seen, there is marked temperature dependence for the methine bridge resonances, H-3B and H-4B, and for the H-5B resonance. This behaviour has been reported previously for 6,8-dinitro-BIPS and has been adequately explained in terms of cis to trans isomerisation about the bond involving the TTC and TTT isomeric forms.9 At 213 K the position of a second component to the H-5B resonance became apparent as a broadened signal at around 8.64 ppm. At 193 K the resonance at 8.64 ppm becomes more resolved and it was observed that saturation of the H-5B resonance at 8.23 ppm also resulted in the complete loss of the new signal at 8.64 ppm. This clearly demonstrates that these two resonances are due to H-5B in isomeric chemical exchange.
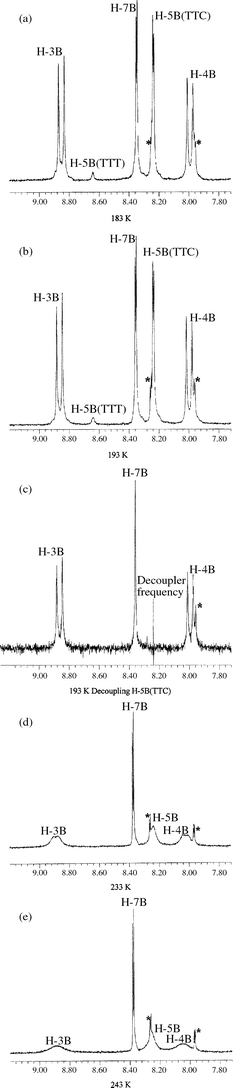 | ||
| Fig. 1 Temperature effects on the 400 MHz 1H NMR spectrum of 6-nitro-8-bromo-BIPS in methylene chloride. From top to bottom at (a) 183 K, (b) 193 K, (c) 193 K saturating the 5H(TTC) resonance demonstrating chemical exchange with the H-5B(TTT) resonance, (d) 233 K and (e) 243 K. Resonances marked with asterisks are due to the spiropyran form. The scale is in ppm. | ||
Using the relative integrals of the TTT and TTC H-5B resonances at either 193 or 183 K, which allow us to estimate the equilibrium ratio at these temperatures, the energy difference between these two isomers was estimated to be 4.6±0.7 kJ mol−1. Unfortunately, with a 400 MHz spectrometer we are limited by the fact that the H-5B resonance is just above the noise level at temperatures approaching the freezing-point of methylene chloride and a higher field spectrometer should give better results since the exchange broadening phenomena will occur at higher temperature.
The isomerisation rate dependence of the H-5B resonance lineshapes was simulated using G-NMR, available from Cherwell Scientific. Comparison of these simulations with our spectra obtained at different temperatures allows us to estimate the exchange rate at the different temperatures. The Arrhenius treatment of these data (Fig. 2, Table 2) yields an energy barrier for TTC–TTT isomerisation of 43.6±3 kJ mol−1 (error derived from linear regression). The error bars were derived from our confidence in the measurement of linewidth and position, but perhaps a much more significant and systematic error, and one that cannot be easily accounted for, is the fact that the true positions of the individual H-5B resonances cannot be assumed to be independent of temperature for such an equilibrated isomeric mixture. Also, equilibrium bond length changes as a function of temperature can shift peak positions. This should be remembered when evaluating this result. Further note that the identity of the TTC isomer as the dominant form has been assumed based on the behaviour of 6-nitro-8-bromo-BIPS in DMSO and on previously obtained evidence on 6,8-dinitro-BIPS and this conformation has not yet been independently determined in methylene chloride.
 | ||
| Fig. 2 Arrhenius plot for the TTC–TTT isomerisation of the 6-nitro-8-bromo-BIPS merocyanine form. | ||
Conclusion
If all assumptions made were correct, then the first experimental estimate of the energetics of the rapid ground state TTC→TTT cis–trans isomerisation about the bond of a spiropyran merocyanine form has been achieved by 1H NMR spectroscopy it is also possible to calculate the rates of TTC→TTT and TTT→TTC interconversion together with the energy barrier and free energy difference between the two isomers.Acknowledgements
We thank the European Commission for funding this work as part of its BRITE EURAM programme, contract No. BRPR-CT96-0328, project No. BE-3380, fellowship No. BRMA-CT 97-5041. We are also grateful to Walter Stringo for his assistance with NMR spectroscopy.References
- F. Wilkinson, D. R. Worrall, J. Hobley, L. Jansen, S. L. Williams, A. J. Langley and P. Matousek, J. Chem. Soc., Faraday Trans., 1996, 92, (8), 1331 Search PubMed.
- H. Takahashi, H. Murakawa, Y. Sakaino, T. Ohzeki and O. Yamada, J. Photochem. Photobiol., A, 1988, 45, 233 Search PubMed.
- H. Takahashi, K. Yoda, H. Isaka, T. Ohzeki and Y. Sakaino, Chem. Phys. Lett., 1987, 140, (1), 90 CrossRef.
- N. P. Ernsting and Th. Arthen-Engeland, J. Phys. Chem., 1991, 95, 5502 CrossRef CAS.
- R. Heligman-Rim, Y. Hirschberg and E. Fischer, J. Phys. Chem., 1962, 66, 2465.
- N. Tamai and H. Masuhara, Chem. Phys. Lett., 1992, 191, (1,2), 189 CrossRef.
- S. Schneider, A. Mindl, G. Elfinger and M. Melzig, Ber. Bunsen-Ges. Phys. Chem., 1987, 91, 1222 Search PubMed.
- T. Yuzawa, K. Ebihara, H. Hiura, T. Ohzeki and H. Takahashi, Spectrochim. Acta, Part A, 1994, 50, (8/9), 1487 CrossRef.
- J. Hobley, V. Malatesta, R. Millini, L. Montanari and W. O'Neil Parker and Jr., Phys. Chem. Chem. Phys., 1999, 1, 3259 RSC.
- R. Guglielmetti, in Photochromism Molecules and Systems, ed. H. Durr and H. Bouas-Laurent, Elsevier, New York, 1990, ch. 8..
- J. Hobley, V. Malatesta and L. Montanari, XVII International IUPAC Conference on Photochemistry, Sitges, Barcelona, Spain, 19–24 July 1998, Book of Abstracts, p. 188..
- J. Keeler, D. Neuhaus and M. P. Williamson, J. Magn. Reson., 1987, 73, 45.
Footnote |
| † Marie Curie Research Fellow 1997–1999. Present address: Advanced Science Research Centre, Japan Atomic Energy Research Institute, 25-1, Mii-Mianami-Machi, Neyagawa, Osaka 572-0019, Japan. E-mail: j.hobley@apr.jaeri.go.jp; Fax:+81720310596; Tel:+81 720 31 0943. |
| This journal is © the Owner Societies 2000 |