π-Cloud and non-bonding or H-bond connectivities in photochromic spiropyrans and their merocyanines sensed by 13C deuterium isotope shifts
Jonathan Hobley†*a, Vincenzo Malatestaa, William Giroldinia and Walter Stringob
aGreat Lakes Chemical Italia, Via Maritano 26, 20097 San Donato Milanese, Italy
bEnitecnologie, Via Maritano 26, 20097 San Donato Milanese, Italy
First published on UnassignedUnassigned22nd December 1999
Abstract
Deuterium induced isotopic shifts in the 13C NMR spectra of spiropyrans and equilibrium mixtures of their TTT and TTC merocyanine form isomers give insights into rovibrational and geometric changes that occur upon isotopic substitution which also result in changes in nuclear shielding. Isotopic shifts in the 13C NMR spectra of C-3 deuterated 1′,3′,3′-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2-indoline], (3D-6-nitro-BIPS), are observed three bonds away from the point of deuteration in this spiropyranic form of the molecule, whereas for C-3 deuterated 1′,3′,3′-trimethyl-6,8-dinitrospiro[2H-1-benzopyran-2,2-indoline], (3D-6,8-dinitro-BIPS), which is stable as a merocyanine the isotope shifts have a range of five bonds. For C-4 deuterated 6,8-dinitro-BIPS (4D-6,8-dinitro-BIPS) the isotope shift range extended to four bonds.
Introduction
The electrocyclic reaction responsible for the photochromism of spiropyrans is shown in Scheme 1, whereby the colourless closed form is transformed, thermally or photochemically, into the strongly coloured merocyanine(s).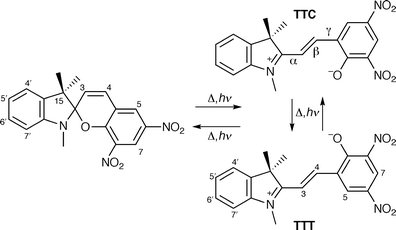 | ||
| Scheme 1 | ||
We have previously shown that the merocyanine form of the spiropyran-6,8-dinitro-BIPS exists in different solvents as an isomeric distribution made up of the TTC and TTT isomers with the TTC isomer dominating.1 We have also observed 1H NMR line broadening effects with this compound that are indicative of rapid rotation about the γ-bond adjacent to the methine bridge.1,2 The activation barrier and ΔH° for this isomerisation were estimated to be 43.6 and 4.6 kJ mol−1 respectively. Further, we reported that the C-3–H bonds of spiropyran merocyanines are labile and undergo isotopic exchange in organic solvents such as methanol, DMSO, acetone and chloroform upon addition of deuterium oxide.1–3 The H–D exchange was slow on the 1H NMR time-scale, taking of the order of 1 day to occur. The lability of the C-3–H bond of these merocyanine isomers may result from a bond polarisation strongly influenced by the zwitterionic charge distribution between the indolinic nitrogen and the phenolate oxygen at C-9. Furthermore, we thought that any non-bonding interaction or H-bonding between this seemingly polar C-3–H bond and the phenolate oxygen at C-9 in the TTC isomeric form would affect the equilibrium merocyanine geometry and conformation, if indeed present. We therefore sought experimental evidence for this conformational perturbation to understand whether there is an interaction between C-3–H and O–C-9. In the present work we mapped the effect of C-3 and C-4 deuteration (the deuterium isotope effect4,5) on 13C NMR spectra of the rapidly exchanging and equilibrated TTT and TTC merocyanine isomers of 6,8-dinitro-BIPS and the C-3-deuterium isotope effect for the spiropyran, 6-nitro-BIPS. In this way we were able to compare the isotope effects on the conjugated merocyanine forms and on the spiropyran form and also the effect of the proximity of the deuteration site to the phenolate oxygen in the merocyanine form.
Deuterium isotopic shifts Δn, where Δ is the shift and n is the number of bonds from the deuteration site, in 13C spectra have recently become a useful tool in understanding the effects of resonance assisted hydrogen bonding (RAHB),6 hydrogen bonding7–9 and extended conjugation of organic compounds.10–12 In order to explain the origin of intrinsic Δns, it is assumed that the Born–Oppenheimer approximation is valid and that deuterium substituted compounds have essentially the same electronic properties as their proton substituted counterparts. Therefore, the intrinsic isotopic shift in 13C NMR spectra are said to originate from the mass difference between the isotopes altering the rovibrational wavefunctions of the ground state and leading to a corresponding small reduction in the C–D bond length compared with that of the C–H bond.4,5 This bond length reduction is small13 and therefore difficult to evidence experimentally because it would be lost within the experimental error of X-ray and neutron diffraction measurements. The magnitude of the deuterium-induced isotopic 13C resonance shift is most pronounced at the site of deuteration and its magnitude generally falls off very rapidly as a function of bond separation between the point of deuteration and the point at which the resonance originates. Whereas the origin of the intrinsic isotope shifts have been suggested to depend on a rovibrational term, the range is said to be dependent upon a electronic term,14 with the π-cloud apparently capable of assisting the long-range transfer of the isotope shift. In fact, it would be hard to distinguish a rovibrational mechanism from an electronic mechanism for this so-called electronic transfer term,14 since the rovibrational wavefunction is inextricably linked to the electronic wavefunction. In summary, rovibrational changes upon deuteration create equilibrium geometric differences in the molecule that become manifest as slight changes in orbital overlaps and or electron density which in turn result in attenuation of nuclear shielding at certain sites in the molecule. In conjugated systems where the molecular orbital is extensive, these attenuations can often be detected far from the centre of deuteration. For the compound shown in Scheme 2, which has such an extended π-system, Δns have been measured for which n extends to 12 bonds away from the deuteration site.10 However, the magnitude of the effect is small (10 ppb or less) for n>2.
 | ||
| Scheme 2 | ||
Where H-bonding or RAHB is implicated, the effects can be transferred through the hydrogen bond via the intrinsic mechanism described above or may result from other mechanisms due to equilibrium perturbations in isomeric conformation or strain induced conformation changes resulting from the H-bond and D-bond strength difference.6–9 These equilibrium H-bonding effects can cause significant geometric changes many bonds away from the centre of deuteration which result in resolvable isotope shifts. Large long range isotope shifts can therefore be a useful indication of geometric modifications occurring within a molecule as a result of H-bonding.
Experimental
6-Nitro-BIPS was obtained from Aldrich and was deuterated at C-3 by dissolving the spiropyran form in chloroform and then layering deuterium oxide above this chloroform solution. Deuterium exchange takes place on the merocyanine form and the deuteration rate therefore depends on the concentration of merocyanine in equilibrium with the closed spiropyran form. The latter will become progressively deuterated over a period of several months through the merocyanine form, with which it is in thermal equilibrium. The C-3 deuterated spiropyran was crystallised from the separated chloroform layer.6,8-Dinitro-BIPS was prepared using normal preparative methods.15 Its purity was established using 1H NMR spectroscopy and thermogravimetric analysis. C-3 deuteration was accomplished in the same way as for the 6-nitro-BIPS except that the time required for complete deuteration was only 1 day since 6,8-dinitro-BIPS exists in chloroform predominantly as the merocyanine form. Further recrystallisation was carried out from dry acetone in a dry-box to avoid D–H exchange at C-3 caused by ambient moisture. C-4 deuterated 6,8-dinitro-BIPS was prepared in the same way as 6,8-dinitro-BIPS, but using 3,5-dinitrosalicylaldehyde with deuteration at the aldehyde group in position 2 on the ring.15 The extent of deuteration was found to be 100% using 1H NMR spectroscopy. 1H and 13C NMR experiments were carried out on a Varian VXR 400 MHz spectrometer in 99.96% DMSO-d6 solution (Cortec).
One-dimensional 13C NMR, of C-3-D/C-3-H isotopomeric mixtures of 6-nitro-BIPS and both C-3-D/C-3-H and C-4-D/C-4-H isotopomeric mixtures of 6,8-dinitro-BIPS, was used to evaluate the D-induced 13C resonance isotope shifts. Using such isotopomeric mixtures very accurate values of isotope shift are obtained since both of the isotopomers are subjected to identical conditions. The assignments of the deuterated and protonated forms were confirmed using mixtures of different D-enrichment levels to give different peak ratios of isotopomers. DEPT,16 HETCOR17 and COLOC18 sequences were used to assign the 13C resonances of 6-nitro-BIPS and 6,8-dinitro-BIPS. Delays optimised for J(CH) of 140 Hz were used for the HETCOR and delays optimised for J(CH) of between 2 and 10 Hz were used for the COLOC experiments.
Results and discussion
13C NMR resonances and assignments are given in Tables 1 and 2 (see also Scheme 3).| Atom | Chemical shift (ppm) | Atom | Chemical shift (ppm) |
|---|---|---|---|
| C-3 | 121.331 | C-2′ | 51.788 |
| C-4 | 128.176 | C-3′ | 135.734 |
| C-5 | 122.697 | C-5′ | 119.320 |
| C-5* | 118.834 | C-7′ | 106.920 |
| C-6 | 140.416 | C-4′, C6′ | 121.437, 127.546 |
| C-7 | 125.605 | C-8′ | 147.307 |
| C-8 | 115.275 | N-methyl | 28.422 |
| C-9 | 159.289 | Geminal methyls | 19.597, 25.607 |
| C-2 | 106.055 | ||
| Atom | Chemical shift (ppm) | Atom | Chemical shift (ppm) |
|---|---|---|---|
| C-3 | 110.813 | C-2 | 51.404 |
| C-4 | 151.737 | C-2′ | 169.440 |
| C-5 | 134.534 | C-3′ | 143.192 |
| C-5* | 126.195 | C-4′, C-7′ | 114.357, 122.628 |
| C-6 | 128.145 | C-5′, C-6′ | 128.388, 128.706 |
| C-7 | 125.223 | C-8′ | 141.804 |
| C-8 | 140.772 | N-Methyl | 33.602 |
| C-9 | 182.409 | Geminal methyl | 25.854 |
For the merocyanine 6,8-dinitro-BIPS, the C-9 carbonyl resonance at 182.4 ppm indicates that the C-9–O bond is partially double bonded. The positions of the C-3 and C-4 resonances of 110.8 and 151.8 ppm, respectively, reflect the alternation of negative and positive charge induced by the terminal indoline nitrogen and oxygen at C-9 on to the methine bridge with C-3 and C-4 assuming a partial δ− and δ+ character, respectively. It is worth noting that in the spiropyran 6-nitro-BIPS these two resonances are separated by just over 7 ppm and the C-3–C-4 bond is therefore much less polarised in the spiropyran form of the molecule. Diagrams summarising the isotope shifts are given in Scheme 3.
 | ||
| Scheme 3 (A) Numbering system used in this study. (B) Magnitudes of isotopic shifts (ppb). If no value is given, an isotopic shift was not resolved. | ||
In the D–H-isotopomer mixtures the 13C resonances experiencing an isotopic shift are split into two separate resonances as shown in Fig. 1. To keep the same terminology as other authors, we define an upfield resonance shift upon deuteration as a positive Δn.6,8,9,14
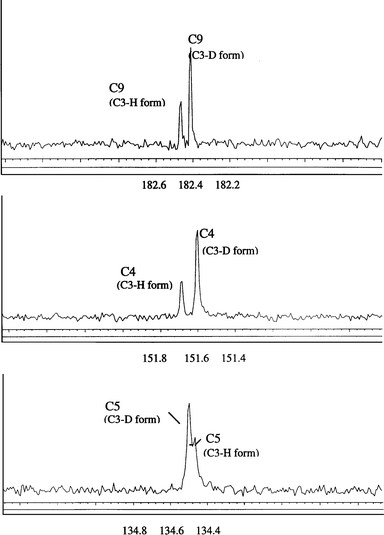 | ||
| Fig. 1 Isotope shifts for a C-3–D/C-3–H isotopomeric mixture observed as split 13C resonances for C-9, C-4 and C-5. Scale in ppm. | ||
In the case of 3D-6-nitro-BIPS the isotope shift behaviour is essentially that of a system with intrinsic rovibrationally induced isotope shifts that are transferred in a relatively unconjugated electronic ground state and there are no noteworthy long range shifts. The cyclic nature of the spiropyran form probably limits the initial size and subsequent transfer of the 3D isotope effect around the pyran ring system owing to its rigid structure. The Δ3 of 15 ppb at C-2′ on the indoline ring may result from an efficient geometric or orbital overlap distortion caused by a sideways directed C-3–D/H bond vibration since the two ring systems are orthogonal to each other and linked by an sp3 spiro bond. The two benzene rings display no shifts reflecting their rigid frame and isolated molecular orbitals.
In contrast, it is apparent that for 4D-6,8-dinitro-BIPS the extent and initial size of the isotope shift is increased with respect to 3D-6-nitro-BIPS, as may be expected on account of the former compound's more extensive conjugation and less rigid structure.
The extent and magnitude of these shifts is explicable in terms of the increase in conjugation alone with transfer of the intrinsic effect described by the electronic term (which itself may be rovibrational or electronic in origin),14 since the shifts clearly follow the path of conjugation and drop off rapidly as the distance from the deuteration site increases.
For 3D-6,8-dinitro-BIPS we see a smaller Δ1 (280 vs. 347 ppb) and a longer range in the resulting Δns than for its 4D counterpart. These Δ1s extend to a measurable Δ5 of 19 ppb at C-8 and large Δ4s of 54 ppb at the C-9 position and −32 ppb at C-5. The magnitude and position of these Δns do not seem consistent with a transfer of the effect via the electronic cloud alone especially in view of the Δns observed for 4D-6,8-dinitro-BIPS. Other workers have observed large long range (n>3) values of Δn for compounds where the isotope substitution site is involved in strong intramolecular H-bonding and have found that H-bonds are effective at communicating the isotope effect.6–9,14 As mentioned previously, three mechanisms of Δn transfer resulting from H-bonding have been identified, namely intrinsic, strain and equilibrium effects.
In view of the long range isotope shifts observed here for C-3 deuterated isotopomer mixtures, we consider the possibility of C-3–H—O–C-9 hydrogen interactions. We consider both steric effects and dipolar interaction, although we must state that we would expect any dipolar interaction to be much weaker than for systems which have been conventionally accepted as H-bonds, involving nitrogen and oxygen with hydrogen, and which have so far been studied using isotope shifts. In the case of 6,8-dinitro-BIPS there is an equilibrium between the TTC and TTT isomers with the TTC isomer dominating. In this TTC conformation the hydrogen at C-3 is in close proximity to the phenolate oxygen at C-9 forming a six-membered ring. Further, this bond is obviously polarised since the proton at C-3 is so easily replaced by deuterium when contacted with deuterium oxide in organic solvents. It is therefore conceivable that in this conformation there is an intramolecular dipolar attraction or weak H-bond between C-3–H and the phenolate oxygen at C-9 and that this interaction provides the mechanism for the observed long range isotope shifts either by chemical equilibrium shift between TTC and TTT or by a shift in equilibrium geometry (e.g., planarity difference) or via an intrinsic mechanism directly from the C-3–D bond to the C-9 phenolate oxygen and hence C-9. Heating the solution to 393 K did not alter the magnitude of the measured shifts, which may indicate that the shifts are not an equilibrium phenomena or may simply be because the equilibrium is not sufficiently affected by this temperature change to register a difference in the isotope shift.
Owing to the apparent sensitivity of the isotope shift to geometric changes and since the equilibrium C-3–H bond rovibrational wavefunctions and hence dimensions are different from those of the C-3–D bond, we cannot rule out the possibility that the transfer of the long range isotope shifts is due to a purely non-bonding steric interaction difference between the C-3–H/C-3–D bonds and the C-9 oxygen and that this difference attenuates the conjugation and π-cloud overlap.
It is clear that for 6,8-dinitro-BIPS some geometric perturbation occurs upon C-3 deuteration extending from the C-3–D bond to C-9 (Δ4 of 54 ppb), possibly intrinsically via the C-9–O bond, which is in close proximity in space to the point of deuteration and/or via equilibrium shifts. Equilibrium and or intrinsically generated geometrical changes at C-9 may be responsible for the Δ5 of 19 ppb that is felt at C-8, since C-8 is nitro substituted and therefore conjugated to the phenolate oxygen at C-9. Significantly, although both of the NO2 groups are five carbon atoms away from the point of deuteration, only the C-8 13C resonance experiences a measurable isotope shift. This is highly suggestive that in some way the oxygen is mediating the transfer of the effect.
The Δ4 at C-5 of −32 ppb may be due to steric strain differences between the C-3 protonated and C-3 deuterated forms. Alternatively, it could be due to a shift in equilibrium between the TTT and TTC isomeric forms. The fact the Δ1 for 3D-6,8-dinitro-BIPS is smaller than that for 4D-6,8-dinitro-BIPS probably results from dissipation of the rovibrational effects in two directions both towards C-3 and the oxygen at C-9 which would effectivly dilute the effect at C-3. This dilution is also seen at C-4 which has a Δ2 of 84 ppb compared with the Δ2 of 146 ppb at C-3 of 4D-6,8-dinitro-BIPS. We feel that it is an important result that for 4D-6,8-dinitro-BIPS, which has the same conjugation as its 3-deuterated counterpart and for which no such non-bonding or H-bond transfer mechanism would be expected, the observed shifts are not similar.
Conclusion
Deuterium isotope shift mapping is a powerful tool in the identification of rovibrational connectivities through the ground state π-system of a molecule.In the present case, isotope shift mapping has been used to demonstrate that the methine bridge C-3–H/D bond of 6,8-dinitro-BIPS is connected with sites on the molecule up to five bonds away. The magnitude and position of these long range effects cannot be explained solely upon the π-delocalisation of the system.
From the present work, we cannot differentiate whether H-bonding or non-bonding interactions result in the long range transfer of the isotope shift in 3D-6,8-dinitro-BIPS, but we feel that both suggested mechanisms have merit and the overall effect is probably a combination of non-bonding repulsion and electrostatic attraction.
Acknowledgements
J.H. thanks the European Commission for funding this work as part of its BRITE EURAM programme, contract No. BRPR-CT96-0328, project No. BE-3380, fellowship No. BRMA-CT 97-5041. J.H. also thanks all of the staff of Great Lakes Chemical Italia for being such good and patient hosts for the duration of his fellowship.References
- J. Hobley, V. Malatesta, R. Millini and L. Montanari and W. O'Neil Parker, Jr., Phys. Chem. Chem. Phys., 1999, 1, 3259 RSC.
- J. Hobley and V. Malatesta, Phys. Chem. Chem. Phys., 2000, 2, 57 RSC.
- J. Hobley, V. Malatesta and L. Montanari, XVII International IUPAC Conference on Photochemistry, Sitges, Barcelona, Spain, 19–24 July 1998, Book of Abstracts, p. 188..
- K. L. Servis and R. L. Domenick, J. Am. Chem. Soc., 1986, 108, 2211 CrossRef CAS.
- C. J. Jameson, J. Chem. Phys., 1977, 66, 4983 CrossRef CAS.
- P. E. Hansen, J. Mol. Struct., 1994, 321, 79 CrossRef CAS.
- W. H. Gmeiner and C. D. Poulter, J. Am. Chem. Soc., 1988, 110, 7640 CrossRef CAS.
- J. Reuben, J. Am. Chem. Soc., 1986, 108, 1735 CrossRef CAS.
- J. Reuben, J. Am. Chem. Soc., 1987, 109, 316 CrossRef CAS.
- S. Berger and H. Kunzer, Angew. Chem., Int. Ed. Engl., 1983, 22, 321 CrossRef.
- Z. Meic, D. Vikic-Topic and H. Gusten, Org. Magn. Reson., 1984, 22, 237 Search PubMed.
- Z. Meic, P. Novak, D. Vikic-Topic and V. Smrecki, Magn. Reson. Chem., 1996, 34, 36 CrossRef CAS.
- C. J. Jameson and H. J. Osten, J. Chem. Phys., 1984, 81, 4293 CrossRef CAS.
- P. E. Hansen, Magn. Reson. Chem., 1993, 31, 23 CAS.
- R. Guglielmetti, in Photochromism Molecules and Systems, ed. H. Durr and H. Bouas-Laurent, Elsevier, New York, 1990, ch. 8. Search PubMed.
- D. M. Dodrell, D. T. Pegg and M. R. Bendall, J. Magn. Reson., 1982, 48, 323.
- H. J. Reich, M. Jautelat, M. T. Messe, F. J. Weigert and J. D. Roberts, J. Am. Chem. Soc., 1969, 91, 7445 CrossRef CAS.
- H. Kessler, C. Griesinger, J. Zarbock and H. Loosli, J. Magn. Reson., 1984, 57, 331 CAS.
Footnote |
| † Marie Curie Research Fellow 1997–1999. Present address: Advanced Science Research Centre, Japan Atomic Energy Research Institute, 25-1, Mii-Mianami-Machi, Neyagawa, Osaka 572-0019, Japan. E-mail: j.hobley@apr.jaeri.go.jp; Fax:+81 720 310596; Tel:+81 720 31 0943. |
| This journal is © the Owner Societies 2000 |