Self assembly of a novel water soluble iron(II) macrocyclic phosphine complex from tetrakis(hydroxymethyl)phosphonium sulfate and iron(II) ammonium sulfate: single crystal X-ray structure of the complex [Fe(H2O)2{RP(CH2N(CH2PR 2)CH2)2PR}]SO4·4H 2O (R = CH2OH)
John C. Jeffery*a, Barbara Odell*b, Nicola Stevens*c and Robert E. Talbot*c
aSchool of Chemistry, University of Bristol, Cantocks Close, Bristol, UK BS8 1TS. E-mail: john.jeffery@bristol.ac.uk
bDyson Perrins Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QY. E-mail: barbara.odell@chemistry.oxford.ac.uk
cAlbright and Wilson UK Ltd., PO Box 80, Trinity Street, Oldbury, West Midlands, UK B69 4LN. E-mail: bob_e_talbot@albriw.com
First published on UnassignedUnassigned6th January 2000
Abstract
The water soluble Fe(II) macrocyclic phosphine complex [Fe(H2O)2{RP(CH2N(CH2PR 2)CH2)2PR}]SO4·4H 2O (R = CH2OH) has been characterised by single crystal X-ray diffraction and is formed by a remarkable self-assembly reaction between iron(II) ammonium sulfate and tetrakis(hydroxymethyl)phosphonium sulfate (THPS).
There is increasing interest in catalytic transformations carried out in aqueous media employing water soluble phosphine metal complexes.1–4 In the latter context, phosphine ligands such as P(CH2OH)3 have been shown to be useful precursors for the synthesis of water soluble transition metal complexes.4 Albright and Wilson have an interest in the related phosphonium salt [P(CH2OH)4]2SO45 because of its effectiveness as a biocide in oil wells. Recently, it was noted that [P(CH2OH)4]2SO4 also aids the dissolution of iron sulfide deposits and particulates. Iron sulfide arises both from anaerobic microbial activity in oil wells and as a result of indigenous H2S; deposits cause flow restrictions in vessels and pipework and particulates can upset oil/water separation and lead to damage. In oil well situations, the dissolution of FeS by [P(CH2OH)4]2SO4 is accompanied by a red coloration of produced water from the well. The aim of this study was to explain how [P(CH2OH)4]2SO4 aids the dissolution of FeS and to identify the origin of the resulting red coloration which it was believed might be due to a water soluble iron complex.
The reactions of [P(CH2OH)4]2SO4 and the related phosphine P(CH2OH)3 with various Fe(II) and Fe(III) salts, e.g. FeS, [Fe(NH4)2(SO4)2], FeCl3, FeCl2 and FeSO4, were investigated to find an iron containing laboratory reagent that would model the red complex formed in the oil wells. Only [Fe(NH4)2(SO4)2] produced a red colour with [P(CH2OH)4]2SO4 or P(CH2OH)3 and it was shown that other alkyl phosphonium salts do not give red complexes. Thus it appears that the formation of red water soluble complexes requires an Fe(II) salt, [P(CH2OH)4]2SO4 or P(CH2OH)3 and, crucially, the presence of ammonium ions. Following optimisation of the reaction conditions it was found that addition of 2 equiv. of [P(CH2OH)4]2SO4 to iron(II) ammonium sulfate in water at room temperature, followed by the slow addition of base (NaOH), so that the pH was maintained between 4.5 and 5.0, immediately produced a deep red solution which gave deep red crystals of [Fe(H2O)2{RP(CH2N(CH2PR 2)CH2)2PR}]SO4·4H 2O (R = CH2OH) 1 on standing at 5 °C for two weeks.
The X-ray crystal structure† of 1 (Fig. 1) reveals a cationic octahedral Fe(II) complex with a remarkable tetradentate phosphine ligand in which alternating phosphorus and nitrogen atoms are linked by CH2 spacers to form an eight-membered macrocyclic ring which functions as a cis bidentate phosphine donor to iron. The two nitrogen atoms carry pendant CH2PR2 groups which occupy trans diaxial sites in the metal coordination sphere. Two molecules of H2O occupy the remaining cis coordination sites at iron. The complex is highly symmetric and has crystallographically imposed twofold symmetry leading to chemically equivalent pairs of axial and equatorial phosphorus sites. The 31P{1H} and 1H NMR spectra‡ of the low spin d6 complex 1 are consistent with the solid state structure being maintained in solution. Thus the 31P{1H} NMR spectrum shows two triplets at δ 20.1 [t, 2J(PP) 53 Hz] and −1.5 [t, 2J(PP) 53 Hz] as expected for the two pairs of chemically inequivalent phosphine sites in the complex.
 | ||
| Fig. 1 Molecular structure of the cation of 1 with hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Fe–O 2.071(2), Fe–P(1) 2.1797(8), Fe–P(2) 2.2408(8), P(1)–C(11) 1.849(2), P(1)–C(1) 1.865(2), N(1)–C(3) 1.458(2), N(1)–C(1) 1.467(2), N(1)–C(2) 1.475(2), P(2)–C(21) 1.836(2), P(2)–C(22) 1.842(2), P(2)–C(2) 1.845(2); P(1)–Fe–P(1A) 80.63(4), P(1)–Fe–P(2) 92.41(2), O–Fe–P(2) 88.66(5), O–Fe–P(1) 98.25(5), O–Fe–O(OA) 82.87(9), P(2A)–Fe–P(2) 174.89(3), O(OA)–Fe–P(1) 178.87(4), C(3)–N(1)–C(1) 113.7(2), C(3)–N(1)–C(2) 113.0(2), C(1)–N(1)–C(2) 113.0(2). | ||
The precise mechanism by which complex 1 is formed is uncertain but it seems likely that the reaction involves a Mannich-like condensation of [P(CH2OH)4]2SO4 with two ammonium ions, with the iron(II) ion acting as a template which controls the formation of the macrocycle and its pendant phosphine arms. When the reaction of [P(CH2OH)4]2SO4 and iron(II) ammonium sulfate with more prolonged base addition was monitored by 31P{1H} it was observed that the triplets associated with 1 gradually diminished with concomitant formation of a new complex 2 characterised by two new triplet resonances at δ −12.0 [t, 2J(PP) = 76 Hz] and −22.8 [t, 2J(PP) 76 Hz]. The latter pattern is very similar to that observed for the complex 1 suggesting a related structure, and although complex 2 has not yet been isolated and fully characterised, we tentatively suggest that it might be a neutral hydroxy 2a or oxy-bridged complex 2b of the type shown (Scheme 1) arising from base induced deprotonation of the coordinated H2O molecules in 1.
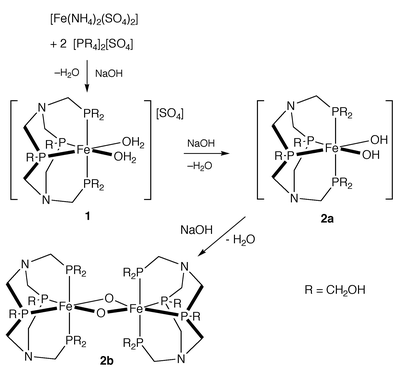 | ||
| Scheme 1 Proposed mechanism for formation of 1 and 2. | ||
This investigation was stimulated by the need to explain how [P(CH2OH)4]2SO4 aids the dissolution of FeS in oil fields leading to a red coloration of the treated water. The speciation of the Fe/S system in natural environments such as oil wells is necessarily complex but our model reactions allow us to tentatively propose that [P(CH2OH)4]2SO4 and NH4+ ions self-assemble iron complexes similar to 1 from FeS that has formed in oil wells owing to sulfate reducing bacteria or indigenous H2S. The key ammonium ions required for the condensation reaction are usually naturally present in oil field waters where [P(CH2OH)4]2SO4 is used. Also, ammonium bisulfite is often added as an oxygen scavenger to oil field injection water used to pressurise oil-bearing formations, thus providing an additional source. The observation that [P(CH2OH)4]2SO4 and FeS do not appear to react in the absence of NH4+ ions, but immediately give red solutions on addition of NH4+, adds further support for the proposed mechanism.
Whilst it was not the original intention of this study to prepare new water soluble catalysts, the novel tetradentate macrocyclic phosphine ligand which has been prepared has obvious potential in this context because it imposes facial octahedral coordination whilst leaving two cis sites free for potential catalytic transformations. Moreover, the ability of self assembled phosphine ligands of this type to strongly bind transition metals suggests that such systems might have a role to play in waste clean-up procedures. The chemistry involved in the self-assembly of the new Fe complex 1 is novel and a patent has been raised to protect its potential applications.1
The authors would like to thank Gary Woodward (Albright & Wilson UK Ltd) and Martin Murray (School of Chemistry, University of Bristol) for their help in spectral interpretation.
References
- UK Pat., HB1103, September 8, 1998..
- L. Higham, A. Powell, M. Whittlesley, S. Wocadlo and P. Wood, Chem. Commun., 1998, 1107 RSC.
- D. J. Darensbourg, F. Joo, M. Kannisto, A. Katho, J. H. Reibenspies and D. J. Daigle, Inorg. Chem., 1994, 33, 200 CrossRef CAS.
- J. W. Ellis, K. N. Harrison, P. A. Hoye, A. G. Orpen, P. G. Pringle and M. Smith, Inorg. Chem., 1992, 31, 3026 CrossRef CAS.
- Albright & Wilson Ltd, UK Pat., 2,145,708B, 1983..
- SHELXTL 5.03 program system, Siemens Analytical X-ray Instruments, Madison, WI, 1995..
- SADABS, A program for absorption correction with the Siemens SMART system, G. M. Sheldrick, University of Göttingen, 1996..
Footnotes |
| † Crystal data for 1: C12H42Fe N2O16P4S, M = 682.3, monoclinic, space group C2/c, a = 12.106(4), b = 14.103(6), c = 15.032(4) Å, β = 90.68(2)°, U = 2566(2) Å3, Z = 4, Dc = 1.766 g cm−3, F(000) = 1432, μ(Mo-Kα) = 0.995 mm−1, R1 = 0.030 [I ≥ 2ς(I)], wR2 = 0.081 for 2912 unique data, 7820 reflections collected (2θ ≤ 55°, 173 K). A full sphere of low temperature data was collected using a Siemens SMART three-circle area detector diffractometer (Mo-Kα X-radiation, graphite monochromator, λ = 0.71069 Å). The structure was solved by direct methods and refined by full matrix least squares on all F2 data using the SHELXTL 5.03 package on a Silicon Graphics Indy computer.6 An empirical absorption correction was applied using SADABS.7 The asymmetric unit contains one half of a molecule of the Fe cation and one half of a disordered SO42− anion both lying astride a twofold axis. There are also two molecules of water of crystallisation. CCDC 182/1498. See http://www.rsc.org/suppdata/cc/a9/a908309j/ for crystallographic files in .cif format. |
| ‡ Selected spectroscopic data for 1: 1H NMR (D2O, 500 MHz) δ 4.63, 4.56[AB, 8H, diastereotopic P(CH2OH)2, 2J(HH) 13 Hz], 4.37 [s, 4H, P(CH2OH)], 3.67 (s, 4H, CH2N), 3.17, 3.10 [AB, 8H, diastereotopic CH2N, 2J(HH) 15 Hz]. 31P{1H} NMR: δ 20.1 [t, 2P, 2J(PP) 53 Hz] and −1.5 [t, 2P, 2J(PP) 53 Hz]. UV–VIS(H2O) λmax 475 nm, (ε = 1496.0 × 10−2 m2 mol−1). Combined yields of 1 and 2 of ca. 88% were estimated by monitoring the conversion of [P(CH2OH)4]2SO4 to products by 31P NMR. Analysis for C12H42FeN2O16P4S: calc.(obs.) C 21.1 (21.3), H 6.2 (6.5), N 4.1 (4.0%). |
| This journal is © The Royal Society of Chemistry 2000 |