The detection of intermediates in the ruthenium(II) catalysed asymmetric hydrogenation of ketones using electrospray ionisation mass spectrometry
Jennifer A.
Kenny
a,
Kees
Versluis
b,
Albert J. R.
Heck
b,
Tim
Walsgrove
c and
Martin
Wills
*a
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: m.wills@warwick.ac.uk.
bDepartment of Biomolecular Mass Spectrometry, Bijvoet Center for Biomolecular Research and Utrecht Institute for
Pharmaceutical Sciences, Utrecht University, Sorbonnelaan 16, 3584 CA, Utrecht, The Netherlands
cSmithKline Beecham Pharmaceuticals, Old Powder Mills, Nr Leigh, Tonbridge, Kent, UK TN11 9AN
First published on 7th January 2000
Abstract
The use of electrospray ionisation mass spectrometry for the detection of the intermediate species involved in the ruthenium(II)/amino alcohol reduction of ketones to alcohols is described.
The use of transfer hydrogenation for the asymmetric reduction of ketones to enantiomerically enriched secondary alcohols has recently been the subject of intense international research work.1 Of the many methods reported for this process, complexes of η6-arene/ruthenium(II) species with β-amino alcohols2 or monotosylated 1,2-diamines3 give some of the very best results in terms of activity and enantioselectivity. In the case of monotosylated diamines the resulting complexes may be employed in conjunction with either PriOH–KOH or formic acid–Et3N as hydrogen source.3 In contrast we have found that the corresponding amino alcohol complexes appear to be compatible only with the PriOH system. In order to gain an insight into the mechanism of the reduction using ruthenium(II) complexes of aminoindanol 1, on which we have recently published, we wished to identify the key intermediates likely to be involved in the process.
Our initial speculation, taking as a starting point the results obtained by Noyori on the analogous monotosylated diamine system, was that the ‘pre-catalyst’ formed by the combination of 1 with ruthenium(II) dimer [RuCl(η6-cymene)]2 was likely to be the 18-electron species 2. Treatment of 2 with base would, we predicted, lead to elimination of HCl to give the 16-electron intermediate 3 which would subsequently be reduced to the 18-electron ruthenium hydride complex 4 by hydrogen transfer from the solvent. In the case of monotosylated diamines Noyori was able to isolate, and characterise through the use of X-ray crystallography, all three intermediates 5–7 analogous to 2–4.4 Despite a number of efforts on our part, we have as yet been unable to obtain crystalline derivatives of any of our speculated intermediates, possibly due to lack of stability relative to the monotosylated diamine variants.

An alternative approach to this problem was, we considered, through the use of electrospray ionisation mass spectrometry (ESI-MS). This technique has been employed recently for the determination of intermediates in a number of catalytic processes, including the oxidative coupling of arylboronic acids5 and a study of the aggregation states of organocopper complexes in solution.6 In the event, this proved to be a highly valuable approach, as described below.
In our first experiment, we prepared a 0.1 M solution of our speculated ‘pre-catalyst’ 2 by combining a slight excess of cis-aminoindanol 1 with ruthenium(II) dimer [RuCl(η6-cymene)]2 in PriOH solution.7 ESI-MS 8 of the resulting solution diluted to 25 mM concentration gave a clean spectrum showing an isotope cluster centred around m/z = 420 (Fig. 1). The masses observed for this cluster and the intensity ratio of the different isotope peaks are in excellent agreement with the calculated pattern for the protonated expected molecule C19H25NORuCl.
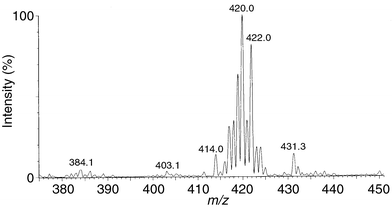 | ||
| Fig. 1 ESI-MS of complex 2 in PriOH. | ||
In the next step of our investigation we added ca. 2 equiv. of an PriOH solution of KOH to the initial solution of 2. After a short period of time (5–10 min) at room temperature, this solution was again examined by ESI-MS by direct injection into the instrument. The resulting spectrum (Fig. 2) showed clearly, in addition to the isotope cluster of compound 2, a new isotope cluster with the isotope at m/z 386 as the highest signal, but starting at m/z 378. The shape of this isotope cluster revealed that both expected molecules 3 and 4 were present. Fig. 3 shows a comparison between the experimentally observed intensities in this cluster and the calculated intensities when it is assumed that the protonated forms of the products 3 and 4 were present at a ratio of 1 to 3. This indicates that the reduction of compound 3 to 4 is likely to be faster than the production of 3 from 2. The less abundent isotope clusters observed around m/z 427 and 444 may be explained by assuming that PriOH can also react with compound 3 (m/z 444) followed by loss of ammonia (m/z 427).
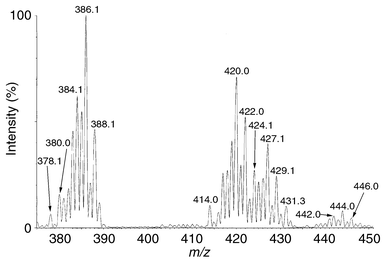
 | ||
| Fig. 3 Comparison of calculated vs. experimental ESI-MS spectra for a mixture of 3 and 4. | ||
In an attempt to obtain evidence for the reduction of 3 by PriOH we repeated the above reaction but with the use of CH2Cl2 as solvent. An ESI-MS spectrum was recorded shortly after the addition of base (Fig. 4) in which a strong set of peaks centred at m/z 384 was observed. The masses and the measured isotope signal intensities correspond to the mass predicted for the compound 3. In the CH2Cl2 solution no suitable hydride source exists for reduction of 3, therefore its lifetime is longer in this solvent.

In a final revealing set of studies, samples were taken of a solution of 4 in PriOH (formed by addition of base) over a period of 90 min following addition of a quantity of acetophenone, which was reduced during this time. Remarkably, species 3 was observed to persist in solution, together with a quantity of unreacted 2, but 4 was not observed. This observation is somewhat at odds with the result illustrated in Figs. 2 and 3, which suggested that 3 was shorter lived than 4. Clearly the presence of ketone (and alcohol reduction product) has an effect of the equilibrium of the reaction.
In view of the results described above, and the lack of evidence for any complex of reduction product (1-phenethanol) with ruthenium, we believe that the hydrogen transfer mechanism probably proceeds via a 6-centre transition state proposed by Noyori for the corresponding reactions of monotosylated diamine complexes of ruthenium(II) (Fig. 5).5 Whilst caution should be exercised in the interpretation of any transition state through inferences from observed intermediates, particularly since the method provides no information on the relative configurations of chiral centres in the complexes, we would consider that our evidence thus presented represents a solid foundation on which to build further studies of the synthetically important process of asymmetric transfer hydrogenation.
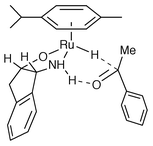 | ||
| Fig. 5 Mechanism of hydrogen transfer. | ||
We thank the EPSRC and SmithKline Beecham Pharmaceuticals for support of a CASE studentship (to J. A. K.) and the British Council for a UK–Dutch Joint Scientific Research Programme Travel Grant.
References
- M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045 CrossRef CAS; R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- (a) (a) J. Takehara, S. Hashiguchi, A. Fujii, S. Inoue, T. Ikariya and R. Noyori, Chem. Commun., 1996, 233 RSC; (b) M. Palmer, T. Walsgrove and M. Wills, J. Org. Chem., 1997, 62, 5226 CrossRef CAS; (c) D. A. Alonso, D. Guijarro, P. Pinho, O. Temme and P. G. Andersson, J. Org. Chem., 1998, 63, 2749 CrossRef CAS; (d) J. A. Kenny, M. J. Palmer, A. R. C. Smith, T. Walsgrove and M. Wills, Synlett, 1999, 1615 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521 CrossRef CAS; S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562 CrossRef CAS; K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738 CrossRef CAS; K. Püntener, L. Schwink and P. Knochel, Tetrahedron Lett., 1996, 37, 8165 CrossRef.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285 CrossRef CAS.
- M. A. Aramadia, F. Lafont, M. Morena-Manas, R. Pleixats and A. Roglans, J. Org. Chem., 1999, 64, 3592 CrossRef.
- B. H. Lipshutz, J. Keith and D. J. Buzard, Organometallics, 1999, 18, 1571 CrossRef CAS.
- The catalyst solutions, with the exception of the CH2Cl2 solutions, were prepared in an analogous manner to those used in the transfer hydrogenation reactions described in ref. 2(b)..
- Mass spectra were acquired on a Micromass Platform single quadrupole instrument. Samples were introduced by loop injection at concentrations of 25 mM..
| This journal is © The Royal Society of Chemistry 2000 |