Photoactivated DNA cleavage via charge transfer promoted N2 release from tris[3-hydroxy-1,2,3-benzotriazine-4(3H)-one]iron(III)
Tucker D. Maurera, Brian J. Krafta, Susan M. Latoa, Andrew D. Ellingtonb and Jeffrey M. Zaleski*a
aDepartment of Chemistry and Biochemistry, Indiana University, Bloomington, IN 47405, USA.. E-mail: zaleski@indiana.edu
bDepartment of Molecular Biology, University of Texas, Austin, TX 78712, USA
First published on UnassignedUnassigned6th January 2000
Abstract
Visible wavelength ligand-to-metal (LMCT) activated N2 release from tris(3-hydroxy-1,2,3-benzotriazine-4(3H)-one]iron(III) produces localized ligand radical intermediates capable of cleaving DNA and represents a new chemical approach to photonuclease design for biological applications.
The antibiotic 6-diazo-5-oxo-L-norleucine utilizes a terminal diazo unit to generate unimolecular diradical intermediates following thermal incubation and loss of N2.1 The kinamycin antibiotics also possess the reactive terminal diazo unit,2 and UV photolysis of kinamycin analogs is thought to generate diradical intermediates that effect DNA cleavage.3 Bioorganic chemists have incorporated this strategy into the design of synthetic photonucleases by preparing diazene and benzotriazole agents that produce diradical intermediates capable of cleaving DNA upon UV excitation.4,5 Although effective, the ability to promote diradical formation using visible region excitation would have significant advantages for in vivo photodynamic therapy applications owing to the increased optical penetration depth by longer wavelengths.6 To this end, we have recently initiated efforts toward developing novel transition metal diazo compounds that use the metal center to activate ligands for N2 release upon optical excitation into metal–ligand charge transfer transitions typically occurring in the visible spectral region. Application of the resulting radical intermediates to DNA cleavage provides approach toward the development of self-contained unimolecular photonucleases for biological applications.
Photochemical activation of diazo compounds occurs via UV
irradiation of the 1(n–π*) transition which
leads to extrusion of N2 and formation of radical
intermediates.7 However, diazo compounds are
also unstable to chemical and electrochemical oxidation and rapidly release
N2 as a reaction product.8–10 We have chosen to exploit this property by
preparing Fe(III) complexes with ligands that posses the
reactive –N![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N– subunit. Fe(III) is a powerful
excited state oxidant11 and is
thermodynamically potent to activate these ligands for N2
release.
N– subunit. Fe(III) is a powerful
excited state oxidant11 and is
thermodynamically potent to activate these ligands for N2
release.
The compound 3-hydroxy-1,2,3-benzotriazine-4(3H)-one 1 (Aldrich) contains the target N2 subunit, and in its deprotonated form, the 3-hydroxy-4-one functionalities strongly chelate Fe(III).12 Reaction of 1 with 3 equiv. of Fe(NO3)3·9H2O in THF–Et3N yields the tris-(chelate 2 as a red powder in nearly quantitative yield.13 The electronic absorption spectrum of 1 in acetonitrile exhibits pronounced π–π* transitions in the 300 nm region and a shoulder at 325 nm corresponding to the forbidden n–π* transition of the diazo unit.7 Photolysis of 1 at λ ≥ 345 nm yields a triplet radical EPR signal (5 K, EtOH) with the signature half-field transition at 1700 G, as well as copious N2 evolution as detected by GC–MS, reflecting the propensity for diradical formation via N2 loss from this organic framework. The absorption spectrum of 2 in the same solvent (Fig. 1) reveals three distinct bands corresponding to a ligand-centered transition (λmax = 300 nm), and two moderately intense O→Fe ligand-to-metal charge transfer (LMCT) bands (λmax = 340, 425 nm) similar to those of Fe(III) tris-catecholates.14
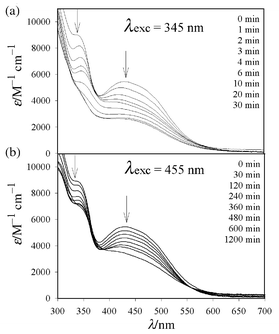 | ||
| Fig. 1 Electronic absorption profiles for anaerobic photolyses of 0.1 mM acetonitrile solutions of 2 at (a) λ ≥ 345 nm and (b) λ ≥ 455 nm. | ||

Anaerobic photolyses of 2 were performed in acetonitrile (0.1 mM) at λ ≥ 345 and 455 nm and monitored with UV–VIS spectrophotometry (Fig. 1). In both cases, rapid photobleaching of the optical spectra are observed upon LMCT excitation of the complex, with partial recovery upon exposure to O2, consistent with formation of Fe(II) in solution. This is confirmed by the disappearance of the rhombic Fe(III) EPR signal at g = 4.3 (Fig. 2) following variable time photolyses at 20 °C. Both reactions exhibit first-order kinetics at early photolysis times, with the reaction at λ ≥ 345 nm (kobs = 9.3 × 10−2 min−1) proceeding considerably more efficiently than photolysis at λ ≥ 455 nm (kobs = 4.0 × 10−3 min−1) under identical experimental conditions. In addition to excitation of both LMCT bands with λ ≥ 345 nm, the disparate reactivity can be attributed to the greater quantum yield for decay of the starting compound upon excitation into the higher energy transition (phi365 = 2.1 × 10−4cf. phi436 = 3.6 × 10−5). This results in simultaneous excitation of both the LMCT and ligand centered n–π* transitions of the Fe(III) compound. To verify LMCT activation of the triazine ligand, photolysis of 1 was conducted in acetonitrile with λ ≥ 455 nm and yielded no reactivity over a 12 h period indicating that photoactivation of 2 at lower energies derives solely from LMCT excitation. Furthermore, reactivity of 2 has been demonstrated at wavelengths up to 500 nm, effectively ruling out the requirement for participation by higher energy excited states in the photoactivation of the triazine ligand.
 | ||
| Fig. 2 Disappearance of the high spin Fe(III) EPR signal of 2 at g = 4.3 as a function of photolysis (λ ≥ 345 nm) time. Relative spin quantitations are denoted above each trace. | ||
To correlate the observed reactivity with N2 extrusion, wavelength dependent photolyses (λ ≥ 345 and 455 nm) were performed on 800 μL solutions of 15 mM 2 under argon in degassed benzonitrile. From headspace GC–MS analysis, N2 production was gauged by comparison of the N2/O2 ratios above the photolyzed solutions relative to unphotolyzed controls. The average of four trials at each wavelength produced an increased N2 content of 112% at λ ≥ 345 nm and 9.7% at λ ≥ 455 nm).15 Together, these results demonstrate that the overall strategy to induce radical formation from charge transfer excited states is indeed operative.
Electronic structure studies of Fe(catecholate)33− have shown that the LMCT excited state is best described as a charge-separated Fe2+–ligand radical. The energy required to photochemically produce a Fe2+–L°+ state can be estimated by the sum of the redox potentials for the free ligand and the metal center, where ΔE = −Eox(1) + Ered(2). The cyclic voltammogram (CV) of 1 demonstrates an irreversible oxidation wave at a peak potential of +1.7 V vs. Ag/AgCl [Eox(1)], derived from rapid denitrogenation.8–10 The CV of 2 exhibits a reversible Fe(III)/(II) redox couple with a half potential of −0.3 V vs. Ag/AgCl [Ered(2)]. Therefore, the minimum energy required to produce the charge-separated excited state, ΔE, is ca. −2.48 eV, or 500 nm from the ground state. However, the energy of this state is exergonic with respect to LMCT excitation at λ ≥ 455 nm and is therefore consistent with charge transfer induced activation and denitrogenation of the triazine ligand at these wavelengths.
The ligand radical intermediate produced upon LMCT excitation of 2 is an effective DNA photocleaving agent. Cleaving ability (%) was determined by quantitating the effectiveness in converting circular supercoiled plasmid DNA (form I) to nicked (form II) and linear DNA (form III) following subtraction of background cleavage due to photolysis of DNA alone (35%). Fig. 3 illustrates the agarose gel electrophoresis of photolysis products of 300 μM 2 in the presence of pUC 118 plasmid DNA (30 μM/bp where bp represents a base pair). Solutions were irradiated anaerobically for 12 h in 1:9 DMSO–Tris buffer (20 mM, pH 7.55) at λ ≥ 400 nm.16 Photolysis produced a mixture of linear (38%) and nicked (27%) forms (lane 3), while thermal incubation effected no DNA cleavage (lane 4). As Fig. 3 shows, the relative amount of cleavage by 2 is significant as photolysis of both plasmid alone (lane 5) and 900 μM free ligand (lane 6) yield identical amounts of nicked DNA (35%), indicating only background levels of DNA photodegradation. Our results demonstrate that although 2 did not generate exclusively linear DNA, it is the only species in Fig. 3 to produce the linear form and consume 100% of the supercoiled form. Additionally, the absence of O2 from the reaction and the presence of the hydroxy radical scavenger DMSO in the buffer effectively rule out participation by O2-derived radicals in the cleavage reaction and implicate a ligand-centered radical as the key intermediate.
 | ||
| Fig. 3 Photoinduced DNA-cleavage of 30 μM/bp pUC 118 by 300 μM 2 following 400 nm photolysis for 12 h at 20 °C (2% agarose gel). Form I: supercoiled plasmid. Form II: nicked plasmid. Form III: linear plasmid. Lane 1: supercoiled DNA; lane 2: linear DNA from EcoRI digest; lane 3: DNA + 2 + hν; lane 4: DNA + 2, no hν; lane 5: DNA + hν; lane 6: 900 μM 1 + DNA + hν. | ||
In conclusion, the above studies describe the preparation and photoreactivity of a novel transition metal triazine compound. The overall strategy of metal complex activation via LMCT excitation in the visible spectral region is operative. Irradiation of the Fe(III) complex in the presence of plasmid DNA affords both single- and double-strand cleavage, with significantly greater efficiency than photolysis of free ligand in threefold higher concentration. From a mechanistic perspective, detection of Fe(II) as a reaction product raises important questions concerning the mode of activation of the kinamycins and 6-diazo-5-oxo-L-norleucine. Are these antibiotics redox activated, and if so, are diradicals or radical ions responsible for the DNA-cleaving reactivity of these agents? Further studies designed to probe the specific nature of the intermediates as well as modulate the reactivity of this system are ongoing.
The generous support of the American Cancer Society (RPG-99-156-01-C) and the Donors of the Petroleum Research Fund (PRF#33340-G4), administered by the American Chemical Society, are gratefully acknowledged.
References
- K. Hiramoto, T. Fujino and K. Kikugawa, Mutat. Res., 1996, 360, 95 CAS.
- S. J. Gould, N. Tamayo, C. R. Melville and M. C. Cone, J. Am. Chem. Soc., 1994, 116, 2207 CrossRef CAS.
- B. G. Maiya, C. V. Ramana, S. Arounaguiri and M. Nagarajan, Bioorg. Med. Chem. Lett., 1997, 7, 2141 CrossRef CAS.
- T. M. Bregant, J. Groppe and R. D. Little, J. Am. Chem. Soc., 1994, 116, 3635 CrossRef CAS.
- S. M. Touami, C. C. Poon and P. A. Wender, J. Am. Chem. Soc., 1997, 119, 7611 CrossRef CAS.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, The Benjamin/Cummings Publishing Company, Inc., Menlo Park, 1978. Search PubMed.
- J. Adamson, D. L. Forster, T. L. Gilchrist and C. W. Rees, Chem. Commun., 1969, 221 RSC.
- R. J. Kobylecki and A. McKillop, 1,2,3 Triazines in Advances in Heterocyclic Chemistry, vol. 19, ed. A. R. Katritzky and J. Boulton, Academic Press, Inc., New York, 1976. Search PubMed.
- V. D. Parker and D. Bethell, J. Am. Chem. Soc., 1987, 109, 5066 CrossRef CAS.
- O. Horvath and K. L. Stevenson, Iron, Ruthenium, Osmium, in Charge Transfer Photochemistry of Coordination Compounds, VCH, New York, 1993, pp. 207–266. Search PubMed.
- R. J. Motekaitis and A. E. Martell, Inorg. Chim. Acta, 1991, 183, 71 CrossRef CAS.
- Compound afforded satisfactory analysis. Calc. for FeC21H12N9O6·0.5 H2O; Fe 10.13; C 45.75; H 2.37; N 22.87. Found: Fe 10.16; C 46.01; H 2.36; N 22.95%. Positive ion ESI-MS match of m/z 543.2..
- T. B. Karpishin, M. S. Gebhard, E. I. Solomon and K. N. Raymond, J. Am. Chem. Soc., 1991, 113, 2977 CrossRef CAS.
- Photolyses were performed for 12 h at λ ≥ 345 nm and 72 h at λ ≥ 455 nm..
- Photolyses were performed at λ ≥ 400 nm owing to the solvatochromatic blue shift of the LMCT transitions of 2 in water/buffer..
| This journal is © The Royal Society of Chemistry 2000 |