DOI:
10.1039/A904925H
(Paper)
Analyst, 2000,
125, 157-162
A glucose biosensor with enzyme-entrapped sol–gel
and an oxygen-sensitive optode membrane†
Received 21st June 1999, Accepted 8th November 1999
First published on UnassignedUnassigned7th January 2000
Abstract
An optical biosensor for the continuous determination of
glucose in beverages based on the canalisation of glucose oxidase into a
sol–gel is presented. The enzyme was entrapped within a glass matrix
by the sol–gel method. The matrix was ground to a powder form and
packed into a laboratory-made flow cell. This minireactor was positioned in
a spectrofluorimeter connected to a continuous sample flow system. An
oxygen-sensitive optode membrane was fabricated from
tris(4,7-diphenyl-1,10-phenanthroline)ruthenium(II) didodecyl
sulfate adsorbed on silica gel particles and entrapped in a silicone-rubber
film. The membrane was situated against the wall of the flow cell to sense
the depletion of oxygen content upon exposure to glucose. The change of the
luminescence intensity of the optode membrane can be related to glucose
concentration. The effects of temperature and pH on the response of the
biosensor were investigated. Storage, stability and repeatability of the
biosensor were also studied in detail. The analytical range of the
biosensor was from 0.06 to 30 mmol dm−3 glucose and the
time taken to reach a steady signal in a flowing solution was 5–8
min. The detection limit was found to be 6 μmol dm−3.
Common matrix interferents such as fructose, galactose, lactose, raffinose,
rhamnose, stachyose, sucrose and other components in beverage samples
showed no interference. The glucose biosensor has been successfully applied
to the determination of glucose contents of beverage samples.
Introduction
In the past two decades, huge efforts in academe and in industrial
laboratories have been devoted to developing biosensors for an array of
analytes.1 However, after the expenditure of
an enormous amount of effort, only a few biosensor systems has been
successfully commercialised. The growing demand for a more practical and
reliable test has been spurring on the development of biosensor devices. In
the clinical diagnosis and food industries, a glucose sensor is considered
to be very desirable for analysis and process control. Clark reported the
first successful electrochemical biosensor using immobilised glucose
oxidase in conjunction with an oxygen electrode over 40 years ago.2 Since then, the literature reporting glucose
biosensors has been accumulating at a very high rate. The majority of them
are electrochemical glucose biosensors. Among them, the most famous
practical device for the determination of blood glucose content was
developed by Yellow Springs Instruments in the early 1970s.3 Most of the electrochemical biosensors are based
on measuring H2O2 (which is generated from the
oxidation of glucose by oxygen) or the response to other electroactive
species (which are previously added to the system and react with the
product of the reaction between oxygen and glucose). These biosensors are
very sensitive to the presence of glucose. However, a very complicated
biosensor system is often specifically designed to overcome the
interferents in different biological samples. The components of the sample
that would be tested have to be known initially in order to correct any
error in measurement. The serious drawbacks of these types of devices are
that the sensing layer is so delicate that it has to be regenerated
frequently; and the weak electrical signal of the device cannot withstand
electric and magnetic interferences, especially in a harsh working
environment.A sol–gel matrix has been proved to be a very useful solid support
for the immobilisation of enzymes as it can retain the enzyme activity and
is considered to be the best way of immobilising glucose oxidase to
date.4 Unfortunately, electrochemical-based
biosensors are not easy to adapt to the sol–gel techniques for the
fabrication of their glucose sensing layer because the sol–gel matrix
does not provide good electrical conductivity.
Optical biosensors have been developing very rapidly since the
mid-1970s.5 The promising features of these
devices are often related to simple sensor design, easy operation, freedom
from electric and magnetic interference and suitability for in
situ or on-line remote monitoring. However, most optical biosensors
developed so far are not as sensitive as the electrochemical biosensors. In
addition, they also suffer from interference from some species in
biological samples. This drawback makes the optical biosensor device very
complicated in design in order to reduce the effects of interferences.
Another major problem is that these biosensors are not robust. As a result,
the real potential of optical biosensors is seldom realised.
For biosensor application, a sol–gel matrix encapsulated with
glucose oxidase has been well studied in recent years.4,6,7 The encapsulated glucose oxidase exhibits
excellent characteristics in terms of activity, lifetime and optical
transparency. In this paper, we report an improved technique for the
fabrication of a sol–gel glucose biosensor. The enzyme was initially
entrapped within a glass matrix by a sol–gel method.4 The matrix was then ground to a powder form and
was packed into a flow cell together with an oxygen-sensitive optode
membrane previously positioned in the flow cell. In this way, a
flow-through system was set up for the determination of glucose. The optode
membrane was fabricated from
tris(4,7-diphenyl-1,10-phenanthroline)ruthenium(II) didodecyl
sulfate [Ru(dpp)3(DS)2] adsorbed on silica gel
particles and entrapped in a thick silicone-rubber film. The change in the
luminescence intensity of the optode membrane can be related to glucose
concentration.8 In this design, the
biosensor exhibited an extremely long lifetime and showed high sensitivity
to glucose. Other favourable attributes of our work are (1) the development
of a very sensitive and extremely stable optical oxygen transducer, (2) the
fabrication of highly active glucose oxidase entrapped in sol–gel
matrix and (3) the assembly of a more practicable flow-through bed system
consisting of sol–gel powder with an almost interference-free
biosensor system. The effects of temperature and pH on the response of the
glucose biosensor were investigated. The properties of storage, stability,
repeatability, response time and interference of the biosensor were also
studied in detail. We successfully applied the proposed method to determine
the glucose content of some beverage samples.
Experimental
Materials
Glucose oxidase (EC 1.1.3.4. from Aspergillus niger) with a
specific activity of 25 000 units per gram of solid, glucose standard
solution (0.10 g cm−3) and β-D-glucose
were obtained from Sigma (St. Louis, MO, USA),
4,7-diphenyl-1,10-phenanthroline, ruthenium(II) chloride
pentahydrate and tetraethyl orthosilicate (TEOS) from Aldrich (Milwaukee,
WI, USA) and sodium dodecyl sulfate (SDS) from Riedel-de Häen (Seelze,
Germany). Tris(4,7-diphenyl-1,10-phenanthroline)- ruthenium(II)
didodecyl sulfate dye ion pair [Ru(dpp)3(DS)2] was
synthesised and purified as described in the literature.9 Silica gel particles (60 Å, 50 μm) were
obtained from Matrex (Merck, Darmstadt, Germany). The one part silicone
sealant SELLEYS (Selleys Chemical, Padstow, NSW, Australia), was purchased
from a local supermarket. All other reagents were of analytical-reagent
grade and used without further purification. The buffer solution for
preparing glucose standards was 0.05 mol dm−3 sodium
phosphate solution (pH 7.0). All solutions were prepared with de-ionised
(DI) water.Preparation of oxygen-sensitive optode membrane
A 50 mg amount of Ru(dpp)3(DS)2 was dissolved in
10 cm3 of acetone and 50 cm3 of ethanol. This
solution was mixed with 2.0 g of silica gel particles, stirred for 2 h at
40 °C, cooled and filtered. The silica gel particles were washed with
60 cm3 of DI water three times, then dried for 6 h at 110
°C. A 0.1 cm3 portion of this oxygen indicator adsorbed on
silica gel was mixed thoroughly with about 0.3 g of silicone sealant. By
the spreading method the mixture was stuck tenaciously to the surface of a
glass plate or a transparent film to form a silicone-based oxygen-sensitive
film. It was left at 55 °C for 24 h to cure. The thickness of the
oxygen sensing layer was estimated to be approximately 100 μm.Preparation of enzyme-doped silica gel powder
A 10.4 g amount of TEOS, 1.8 g of water, 4.6 g of ethanol and 30
mm3 of 0.1 mol dm−3 HCl were mixed and then
stirred using a magnetic stirrer at room temperature for about 8 h to
prepare a clear stock sol–gel solution. A 3.0 cm3 aliquot
of the stock sol–gel solution was placed in a small vial and stirred
under vacuum for 20 min in order to evaporate most of the ethanol. The pH
of the solution was adjusted to about pH 4.5 by adding 20 mm3 of
20 mmol dm−3 sodium phosphate buffer (pH 7.4) (solution
A). In a separate small vial, 18 mg of glucose oxidase and 0.250
cm3 of 20 mmol dm−3 sodium
4-(2-hydroxyethyl)-1-piperazineethanesulfonate buffer solution (pH 7.5)
were mixed, then solution A was added. A vacuum was applied to the stirred
mixture until a gel was formed. The gel was rinsed with 2 cm3 of
water three times. The gel was allowed to dry at 4 °C for 6 d. The
dried gel was collected and ground to a powder form. Unless stated
otherwise, this gel was used for most studies.Assembly of sensing system
The laboratory-made flow-through cell used in this work was machined
from stainless steel and had a chamber volume of approximately 0.45
cm3 (Fig. 1). An oxygen sensing
film plus a blank glass plate were positioned as the window of the flow
cell. The enzyme-doped silica gel powder was subsequently packed into the
flow cell to form a small packed flow bed resulting in a minireactor or
biosensor ready for glucose sensing. This minireactor was situated in a
spectrofluorimeter in conjunction with a continuous sample flow system.
When the glucose biosensor was not in use, it was stored at 4 °C. |
| | Fig. 1 Schematic diagram of the flow-through cell packed with sol–gel
powder and an oxygen optode membrane. (1) Stainless steel cell body; (2)
sol–gel powder; (3) oxygen optode membrane; (4) transparent glass
plate; (5) sample inlet; (6) sample outlet; (7) excitation light beam; and
(8) emission light beam. | |
Instrumentation
Fluorescence intensity was measured on a Perkin-Elmer (Beaconsfield,
Bucks., UK) LS-50B spectrofluorimeter which was controlled by FL WinLab
software. The fluorescence emission intensity at 602 nm was collected at an
excitation wavelength of 460 nm. All measurements were made with 3 nm
bandwidths for both the emission and excitation monochromators. For
gas-phase measurements, oxygen and nitrogen were mixed and flowed
via mass flow controllers [Read Out & Control Electronics 0154
(Brookes Instrument BV, Veenendaal, The Netherlands)] directly to the
sealed oxygen sensing flow-through cell. All measurements were performed in
air-saturated buffer solutions. Using a MasterFlex C/L Model 77120-62
(Cole-Parmer Instrument Co., Chicago, IL, USA) peristaltic pump, the
air-saturated buffer or the air-saturated glucose solutions were pumped
through the flow cell at a typical flow rate of 1.0 cm3
min−1. Unless stated otherwise, all fluorescence
measurements were made under batch conditions at 20 ± 2 °C and
at a pressure of 101.3 kPa.Results and discussion
Response behaviour of oxygen transducer
An oxygen sensing film acting as a transducer was employed to measure
the rate of oxygen consumption in the enzymatic oxidation of glucose. The
optical sensing is based on collision quenching of the fluorescence of
Ru(dpp)3(DS)2 molecules by oxygen molecules.10,11 Hence the biosensor response composed
of a dynamic balance in the diffusion of glucose into the silica gel powder
and oxygen into the silicone-rubber film, and consumption of oxygen in the
enzymatic reaction, resulting in a steady-state decreased oxygen level and,
consequently, an increase in fluorescence intensity. Quenching can be
quantified by intensity quenching measurements. The oxygen quenching
process is described by the well-known Stern–Volmer equation:9,11–13where I is the fluorescence intensity, the subscript 0
denotes the absence of oxygen, K is the Stern–Volmer
constant and pO2 is the partial pressure of oxygen. A
plot of I0/I versus the partial pressure of
oxygen should give a straight line with a slope K and an intercept
of unity on the ordinate. Fig. 2 shows the
curved Stern–Volmer plot of an oxygen sensing film on exposure to
various oxygen concentrations. The curvature was attributed to the
distribution of slightly different quenching environments for the
Ru(dpp)3(DS)2 complex, particularly when quenching
occurs in a solid matrix.9 The emission
spectrum of Ru(dpp)3(DS)2 of the oxygen sensing layer
was very similar to that in the literature.14 The fluorescence intensity showed a very broad
dynamic range for both gaseous and dissolved oxygen measurements, which was
more than 20- and 6-fold, as shown in Fig. 2
and 3, respectively. Certainly, the
sensitivity of response to dissolved oxygen is far better than that in the
literature;11–16 95% of the steady forward and reverse responses
can be reached within 25 s. The concentration of
Ru(dpp)3(DS)2 had a great effect on the fluorescence
intensity. The fluorescence intensity reached a maximum when the amount of
Ru(dpp)3(DS)2 was at 5.2 mg per gram of silica gel
particles. At higher concentrations of Ru(dpp)3(DS)2
the fluorescence intensity decreased significantly, which strongly suggests
that a high content of Ru(dpp)3(DS)2 may cause
self-quenching.9 The compound in the silicone-rubber film was
extremely stable and could be stored for a long period (>1 year) without
any degradation. |
| | Fig. 2 Stern–Volmer curve of the oxygen sensing film at excitation and
emission wavelengths of 460 and 602 nm when subjected to various oxygen
concentrations. | |
 |
| | Fig. 3 Response curves for the oxygen sensing film cycled between (1)
oxygenated water and (2) deoxygenated water. | |
Sol–gel enzyme bed
Proteins entrapped in nanometre-scale cages of sol–gel formed by
the cross-linking of silicone and oxygen units in a sol–gel process
represent a convenient, flexible and efficient immobilisation technique for
enzymes which can retain their biological function in both aged gels and
xerogels. It provides an efficient design that restricts the movement of
the encapsulated recognition molecule and inhibits their intermolecular
interaction but allows free permeation of small analyte molecules.17,18 This study was performed on xerogels.
The glucose oxidase to be encapsulated is added to the sol solution, which
is first subjected to vacuum to remove most of the ethanol because solvents
such as methanol and ethanol have been found to denature enzymes, resulting
in decreased enzymatic activity.The sol–gel procedures are apparently not detrimental to protein
stability and resulted in optically transparent solids that are chemically,
thermally and dimensionally stable immobilised proteins. The enzyme
activation level is easily adjusted simply by changing the amount of
enzyme. One of the main advantages of our biosensor is that the
sol–gel takes the form of powder and it increases the contact surface
area of the biosensor with the solution, which can assist more analytes and
products to diffuse into or out of the sol–gel cages. The powder
packed in a flow-through bed seems to be more suitable for real application
in a flow-through analysis. The packed bed was very stable. It can be
stored for over 10 months at 4 °C without apparently losing the enzyme
activity. Even when kept under ambient conditions for 5 months, the enzyme
activity does not decrease by more than a few per cent.
Dynamic range
The sensing scheme includes the use of glucose oxidase, an enzyme
catalysing the oxidation of glucose by the dissolved oxygen in the
analytical solution. Oxygen has a relatively low solubility in water, the
concentration of oxygen in water in equilibrium with air being only 9.2 ppm
at 20 °C and standard atmospheric pressure.11 The changes in pO2 are
detected via quenching of the fluorescence intensity with the
oxygen sensing film. The decrease measured in the oxygen partial pressure
when glucose is oxidised by the enzyme gives an indirect indication of the
glucose concentration. The fluorescence intensity, due to oxygen
consumption, hence increases and reaches a plateau, the variation being
proportional to the glucose concentration over a wide range. The magnitude
of the analytical signal of the glucose biosensor is therefore determined
by the oxygen quenching constant, the oxygen concentration and the glucose
concentration inside the oxygen sensing membrane. The response behaviour of
the oxygen sensor, the concentration of oxygen in the analytical solution,
the amount or activity of glucose oxidase in the sol–gel powder, the
temperature of the biosensor system and the flow rate of analytical
solution are factors which can strongly affect the working range of the
glucose biosensor. A typical calibration curve for this biosensor in a flow
analysis system is displayed in Fig. 4. The
relative signal change is defined as:13| | | Rs =
(Itest)/(Ibaseline) |
(2)
|
where Itest and
Ibaseline represent the detected fluorescence signals
from the biosensor exposed to glucose solution and buffer solution,
respectively. The maximum Rs measured with this device
increases over fivefold when the biosensor was changed from an
air-saturated buffer solution to an air-saturated 30 mmol
dm−3 glucose solution. In terms of both relative signal
change, Rs and detection limits, our glucose biosensor
shows significant improvements over other optical biosensors of this
kind.5,6,8 The most sensitive and
linear working range is 0.06–2.0 mmol dm−3 glucose
(Itest/Ibaseline = 1.0296[glucose]
+ 0.9968; r2 = 0.995). Even though the sensitivity of
the biosensor decreases above 2.0 mmol dm−3 glucose, the
whole working range of the biosensor can still function between 0.06 and 30
mmol dm−3 glucose (Fig. 4)
when a calibration curve has initially been prepared that covers this range
of glucose concentration. The biosensor performance is not very sensitive
to variations in flow rates from 0.8 to 1.2 cm3
min−1. Increasing the flow rate resulted in an increased
glucose saturation concentration. On the other hand, increasing the
activity of the glucose oxidase in the sol–gel powder can lead to a
lower detection limit and a lower glucose concentration saturation signal.
It is noteworthy that the dynamic working range and detection limit of the
biosensor can be modulated by controlling the relative amount of glucose
oxidase entrapped in the sol–gel powder or the amount of
sol–gel packed in the flow cell. It can fit the multiple requirements
of an analytical method. In this work, a highly sensitive method for
determination of glucose has been realised with a detection limit of 6
μmol dm−3 glucose. The lowest detection limit obtained
was 1 μmol dm−3 when the flow system was in the stop
flow mode.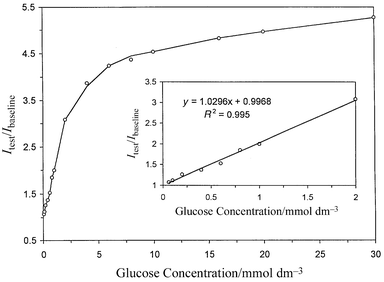 |
| | Fig. 4 Calibration curve for the glucose biosensor at various glucose
concentrations. The inset displays the linear regression curve for lower
glucose concentrations from 0.060 to 2.0 mmol dm−3. | |
Effect of pH
The effect of pH was studied over the range 4.5–8.0. The biosensor
was subjected to a 4.0 mmol dm−3 glucose standard using
phosphate buffer solutions of various pH. The results showed that the
optimum pH value is about 6.0. It was found that pH changes apparently do
not affect the dynamic working range of the biosensor. It is anticipated
that the biosensor will work satisfactorily in the pH range
5.0–8.0.Effect of temperature
It is well known that the analytical performance of both
enzyme-immobilised silica gel and oxygen transducers is highly sensitive to
variations of temperature. The unquenched excited-state lifetimes of
ruthenium(II)–diimine complexes at different temperatures
were 5.8 (0 °C), 5.9 (25 °C), 4.8 (38 °C) and 3.3 μs (60
°C).12 Higher temperatures would result
in a significant decrease in lifetime and fluorescence intensity yield and
also decrease the Stern–Volmer quenching constant of the
ruthenium(II) complex, resulting in a decrease in the
sensitivity of the glucose biosensor. However, raising the working
temperature has a counteracting effect on the biosensor. The activity of an
encaptured enzyme is governed by the kinetics of the enzymatic reaction.
The reaction rate will be increased by raising the working temperature.
Therefore, a study of the effect of temperature on the minireactor was
carried out over the range 15–40 °C. The signal sensitivity
depended strongly on the temperature of the biosensor analytical system, as
expected. The fluorescence intensity changed from 986 to 780 units when in
contact with 100% nitrogen as the temperature increased from 18 to 40
°C whereas the intensity shifted from 48 to 31 units when the
temperature increased from 18 to 40 °C on exposure to 100% oxygen. The
dynamic working range of the biosensor was reduced and the signal
saturation point was reached earlier when the working temperature
increased. However, the response rate of the biosensor increased sharply at
higher working temperatures. The possible reasons are that the enzyme can
acquire higher activity at higher temperatures and subsequently give a more
pronounced signal change with oxygen consumption at a faster rate in the
enzymatic oxidation of glucose. Although the analytical sensitivity was
higher at around 40 °C, for practical purposes temperatures lower than
40 °C are recommended so as to prolong the lifetime of the biosensor
since enzyme can easily be denatured at high temperature.Response time, repeatability and stability of glucose
biosensor
In this study, the response time is defined as the time taken to obtain
a full steady state signal when the biosensor is in contact with an
air-saturated buffer solution and then switches to a known concentration of
air-saturated glucose standard solution. The response time depends on the
enzyme activity, the working temperature, the flow rate of the analytical
solution, the particle size of the sol–gel powder and the thickness
of the oxygen sensing indicator layer. When the oxygen sensor material is
not dispersed into a silicone-rubber film, it can be used to detect gaseous
oxygen with a very fast response time (<1 s). When it is dispersed in a
silicone-rubber film a few micrometres thick, it can be employed to
determine dissolved oxygen in solution with a response time of ≡1
min. As a result, the response time of our glucose biosensor will be mainly
determined by the enzymatic reaction rate of the enzyme and the analyte. A
thicker oxygen indicator layer such as about 100 μm can also be employed
as an oxygen transducer for the fabrication of our glucose biosensor. The
typical time response curve depicted in Fig.
5 exhibits exponential-like behaviour. The response times of the
biosensor were from 5 to 8 min. The relatively short response times are
attributed to the increase in the surface area of the sol–gel powder,
which enhances the exposure of the glucose oxidase to the analyte with a
concomitant effect on the biosensor for reaching a fast steady-state
signal. Moreover, if an enzyme entrapped in the sol–gel matrix has a
higher activity, the response time should also be further shortened. |
| | Fig. 5 Response time and reversibility of the glucose biosensor at excitation
and emission wavelengths of 460 and 602 nm when subjected to various
concentrations of glucose using pH 7.0 phosphate buffers (0.05 mol
dm−3). (1) 0.010; (2) 0.10; (3) 0.50; (4) 1.0; (5) 5.0;
and (6) 10 mmol dm−3. | |
Fig. 5 displays the signal changes of the
biosensor when it is exposed to concentration step changes from 0.010 to 10
mmol dm−3 of glucose in 0.05 mol dm−3
phosphate buffer at pH 7.0. It demonstrates that the biosensor exhibits a
very desirable analytical feature of excellent repeatability although only
two repeats have been performed. The long-term stability of the sensor was
tested over a 280 d period. When the biosensor was stored in a refrigerator
at 4 °C and measured intermittently, the relative signal change of the
biosensor on exposure to 5.0 mmol dm−3 glucose was found
to be above 73% of its initial value over this period. The good stability
of the glucose biosensor can be explained by two possible reasons. First,
most of the positive charges at the channel entrance are balanced by the
negative charges of the nearby acidic residues. Hence there is no
detrimental electrostatic interaction with anionic sites of the
immobilising matrix with the enzyme; as a result, the enzyme is stabilised
upon immobilisation in hydrated silica.19
Second, the cavity in the sol–gel is tailored to the size and shape
of the glucose oxidase. The bottleneck effect of the silica sol–gel
prevents the enzyme from leaking.18–20 The sol–gel powder is so strong that it
can withstand any slightly striking, pressing and liquid flow pressure
without causing cracking and leaching of the enzyme.
Interference test
The interference test was mainly performed in two parts. The purpose of
the first part was to investigate the effects of some common substances on
the quenching response of the oxygen sensing film. It was carried out with
two groups of solutions. The first group of air-saturated solutions
consisted of some potential interferents. The second group of solutions
consisted of some potential interferents together with 2%
Na2SO3. The results showed that there were no
significant signal changes of the oxygen sensing film on exposure to the
interferents. However, caffeine caused a slight interference (Table 1).
Table 1 Effect of potential interferents on the oxygen transducer and glucose
biosensor
| | Signal change for
oxygen transducer | Signal change for
glucose sensor |
|---|
| Interferent | Concentration/ mmol dm−3 | Air-saturated DI water | 2% Na2SO3 solution | No glucose | 1.25 mmol dm−3 glucose |
|---|
| Cetyltrimethylammonium bromide. |
|---|
| Saccharose | 20 | No | No | No | No |
| Fructose | 20 | No | No | No | No |
| Lactose | 20 | No | No | No | No |
| Galactose | 20 | No | No | No | No |
| Raffinose | 20 | No | No | No | No |
| Rhamnose | 20 | No | No | No | No |
| Stachyose | 20 | No | No | No | No |
| NaCl | 100 | No | No | No | No |
| KCl | 20 | No | No | No | No |
| MgSO4 | 5 | No | No | No | No |
| CaCl2 | Saturated | No | No | No | No |
| CuCl2 | Saturated | No | No | No | ≡9% decrease |
| FeCl3 | 2 | No | No | No | No |
| Sodium citrate | 10 | No | No | No | No |
| Lauryl sulfate | 1.5 | No | No | No | No |
| Ascorbic acid | 10 | No | No | No | ≡3% increase |
| CTMABa | 2 | No | No | No | No |
| Vitamin E (in 2 mM CTMABa) | 3 | No | No | No | No |
| Sodium benzoate | 50 | No | No | No | No |
| Caffeine | 2 | No | ≡1% decrease | ≡1% decrease | ≡3% decrease |
The purpose of the second part was to examine the effect of these
interferents on the response of the glucose biosensor. It was evaluated
with two groups of solutions. The first group of solutions contained only
interferents in 0.05 mol dm−3 phosphate buffers. The
second group of solutions consisted of interferents and 1.25 mmol
dm−3 glucose in 0.05 mol dm−3 phosphate
buffers. The results are given in Table
1. It is found that most potential interferents did not give any
significant interference on the response of the glucose biosensor. However,
in the presence of glucose, CuCl2 can give some interference on
the response of the glucose biosensor. It was interesting to find that the
interference from CuCl2 could be diminished from 9 to 4% if the
test solution also contained 10 mmol dm−3 ascorbic acid.
In the presence of glucose, ascorbic acid and caffeine had a slight
interference on the response of the glucose biosensor. However this
interference was reduced to nearly zero if glucose was absent from the
tested solutions. In brief, we were confident that most substances often
found in soft drink samples did not exhibit significant interference on the
determination of glucose using our proposed biosensor method.
Glucose determination in beverage samples
Eight beverage samples of seven brands produced in various places were
bought from local supermarkets and used as our test samples. The pH of a
10.0 dm3 aliquot of each sample solution was adjusted to about
7.0 by addition of a small volume of 0.2 mol dm−3
Na2HPO4 solution. It was then diluted with suitable
phosphate buffer to yield a test sample solution of pH 7.0. The glucose
contents in the samples were determined by the glucose biosensor (Table 2). The relative signal change of each
sample solution was measured and compared with that of a set of glucose
standard solutions. The recovery tests for glucose were performed by adding
various amounts of glucose to the sample solutions. The amounts of added
glucose were then evaluated by using our glucose biosensor. All sample
solutions were air-saturated before testing. The results of the recovery of
the samples are summarised in Table 2.
The recovery tests demonstrate that the glucose biosensor offers an
excellent, accurate and precise method for the determination of glucose in
beverages with almost no effect of interferences from common matrix
substances or components in the beverage sample solution. The glucose
biosensor is characterised by good long-term stability, selectivity, very
high sensitivity, a broad dynamic working range and a relatively fast
response.
Table 2 Results of the glucose assay and the recovery test on beverage
samples
| Beverage (source) | Glucose contenta/ mmol dm−3 | RSD (%) | Glucose added/ mmol
dm−3 | Glucose foundb/ mmol dm−3 | Recovery (%) | RSD (%) |
|---|
| The glucose content is an average of seven tests, determined with the
glucose biosensor. An average of five tests. |
|---|
| Sprite | 61.0 | 3.22 | 50.0 | 48.34 | 96.7 | 2.31 |
| (Hong Kong) | 100.0 | 98.02 | 98.0 | 3.74 |
| 150.0 | 149.2 | 99.3 | 2.98 |
| Sprite | 50.1 | 3.12 | 50.0 | 46.86 | 93.7 | 3.17 |
| (China) | 100.0 | 100.5 | 101 | 2.88 |
| 150.0 | 151.4 | 101 | 4.45 |
| Cola | 185 | 1.32 | 50.0 | 48.86 | 97.7 | 6.83 |
| (Hong Kong) | 100.0 | 101.4 | 101 | 4.74 |
| 200.0 | 201.4 | 101 | 3.31 |
| Pocari Sweat | 72.5 | 1.74 | 50.0 | 51.7 | 103 | 2.06 |
| (Japan, 1998) | 100.0 | 99.1 | 99.1 | 3.21 |
| 200.0 | 199.2 | 99.6 | 2.23 |
| Pocari Sweat | 96.7 | 0.59 | 50.0 | 50.26 | 101 | 2.63 |
| (Japan, 1999) | 100.0 | 100.14 | 100 | 2.59 |
| 200.0 | 198.8 | 99.4 | 3.33 |
| Striker | 153 | 2.21 | 50.0 | 52.44 | 105 | 1.98 |
| (Japan) | 100.0 | 103.7 | 104 | 3.04 |
| 200.0 | 201.4 | 101 | 1.74 |
| Gatorade | 105 | 1.39 | 50.0 | 45.52 | 91.0 | 5.01 |
| (USA) | 100.0 | 97.08 | 97.1 | 1.72 |
| 200.0 | 198.8 | 99.4 | 3.97 |
| Lucozade | 299 | 1.76 | 50.0 | 45.56 | 91.1 | 4.78 |
| (UK) | 100.0 | 97.36 | 97.4 | 3.58 |
| 200.0 | 197.4 | 98.7 | 4.65 |
Acknowledgements
The work described in this paper was partially supported by a grant from
the Research Grants Council of the Hong Kong Special Administrative Region,
China (project No. HKBU 2058/98P).References
- J. Janata, M. Josowicz, P. Vanysek and D. M. DeVaney, Anal. Chem., 1998, 70, 179R CrossRef CAS.
- L. C. Clark Jr., Trans. Am. Soc. Artif. Int. Org., 1956, 2, 41 Search PubMed.
- E. Magner, Analyst, 1998, 123, 1967 RSC.
- Q. Chen, G. L. Kenausis and A. Heller, J. Am. Chem. Soc., 1998, 120, 4582 CrossRef CAS.
- M. C. Moreno-Bondi, O. S. Wolfbeis, M. J. P. Leiner and B. P. H. Schaffar, Anal. Chem., 1990, 62, 2377 CrossRef CAS.
- U. Narang, P. N. Prasad, F. V. Bright, K. Ramanathan, N. D. Kumar, B. D. Malhotra, M. N. Kamalasanan and S. Chandra, Anal. Chem., 1994, 66, 3139 CrossRef CAS.
- S. A. Yamanaka, F. Nishida, L. M. Ellerby, C. R. Nishida, B. Dunn, J. S. Valentine and J. I. Zink, Chem. Mater., 1992, 4, 495 CrossRef CAS.
- W. Trettnak, M. J. P. Leiner and O. S. Wolfbeis, Analyst, 1988, 113, 1519 RSC.
- I. Klimant and O. S. Wolfbeis, Anal. Chem., 1995, 67, 3160 CrossRef CAS.
- J. N. Demas and B. A. DeGraff, Anal. Chem., 1991, 63, 829A CrossRef CAS.
- C. McDonagh, B. D. MacCraith and A. K. McEvoy, Anal. Chem., 1998, 70, 45 CrossRef CAS.
- J. R. Bacon and J. N. Demas, Anal. Chem., 1987, 59, 2780 CrossRef CAS.
- B. D. MacCraith, C. M. McDonagh, G. O’Keeffe, E. T. Keyes, J. G. Vos, B. O’Kelly and J. F. McGilp, Analyst, 1993, 118, 385 RSC.
- R. C. W. Lau, M. M. F. Choi and J. Lu, Talanta, 1999, 48, 321 CrossRef CAS.
- B. Meier, T. Werner, I. Klimant and O. S. Wolfbeis, Sens. Actuators B, 1995, 29, 240 CrossRef.
- E. R. Carraway, J. N. Demas, B. A. DeGraff and J. R. Bacon, Anal. Chem., 1991, 63, 337 CrossRef CAS.
- A. K. Williams and J. T. Hupp, J. Am. Chem. Soc., 1998, 120, 4366 CrossRef CAS.
- B. C. Dave, B. Dunn, J. S. Valentine and J. I. Zink, Anal. Chem., 1994, 66, 1120A CrossRef CAS.
- J. Heller and A. Heller, J. Am. Chem. Soc., 1998, 120, 4586 CrossRef CAS.
- D. Avnir, S. Braun, O. Lev and M. Ottolenghi, Chem. Mater., 1994, 6, 1605 CrossRef CAS.
Footnotes |
| † Presented at the Fifth Asian
Conference on Analytical Sciences, Xiamen University, Xiamen, China,
4–7 May 1999. |
| ‡ Visiting scholar on leave from the College of
Chemistry and Chemical Engineering, Hunan University, Changsha, China. |
|
| This journal is © The Royal Society of Chemistry 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.