Photo-oxidation of 1,3-cyclopentadiene using partially quaternized poly(1-vinylimidazole)-bound ruthenium(II) complexes
Received 23rd August 1999, Accepted 5th November 1999
First published on UnassignedUnassigned22nd December 1999
Abstract
Photo-oxidation of cyclopenta-1,3-diene (CP) with singlet oxygen using tris(2,2′-bipyridine)ruthenium(II) [Ru(bpy)32+] and polymer-bound ruthenium(II) complexes as photosensitizers was investigated in oxygen-saturated ethanol. The polymer-bound ruthenium(II) complexes consist of a bis(2,2′-bipyridine)ruthenium(II) complex coordinated with two imidazolyl residues on partially quaternized poly(1-vinylimidazole) with hexyl groups (C6RuQPIm) and hexadecyl groups (C16RuQPIm) as the alkyl side chains. The photo-oxidation of CP using the polymer photosensitizers effectively occurred in a comparable manner to the Ru(bpy)32+ system: the degree of reaction, being given by the ratio of reacted CP and initial CP concentrations, was high. In particular, all of the CP added was oxidized in the C16RuQPIm system even when the CP concentration was low. This was attributed to the concentration of CP species into the heterogeneous reaction field formed by the polymer
photosensitizers. The Ru(bpy)32+
photosensitizer showed excellent stability and was repeatedly able to be used for the photo-oxidation reaction. During the repeated experiments, the reaction activity for the polymer photosensitizer
systems gradually decreased because the polymer photosensitizers changed to monochloro complexes [CnRu(Cl)QPIm (n=6 or 16)] in which one imidazolyl residue was substituted by a chloride ion. However, these polymer photosensitizers also had excellent stability. Compared with other photosensitizers, such as Rose Bengal and zinc(II) phthalocyaninetetrasulfonic acid, the stability of the ruthenium(II) complex photosensitizers
was excellent.
Introduction
Singlet molecular oxygen, O2(1Δg), plays an important role in both
natural and artificial photochemical processes. The quantum
yields for O2(1Δg) generation have been measured in water
and organic solvents for many photosensitizers.1–8
O2(1Δg)
is believed to be the initial agent in the photodynamic therapy
of cancer (PDT).9–16 Furthermore, in contrast to ground
state molecular oxygen, O2(1Δg) has found considerable utility
since it can undergo photo-oxidation reactions with a
wide variety of electron-rich molecules such as sulfides,17,18
thiols,19,20 phenols21–23 and other organic compounds.24–29One of the most reactive substrates for the endoperoxide
formed by the photo-oxidation reaction is cycloalka-1,3-dienes,
and many studies have been reported using various photosensitizers
such as Rose Bengal, Methylene Blue, chlorophyll,
porphyrins and phthalocyanines, which have a strong absorption
in the visible region.30–37 Some peroxides obtained
in the photo-oxidation of 1,3-dienes with singlet oxygen
are of interest either as pharmaceuticals (e.g. ascaridole30)
or as model compounds for biochemical studies
(e.g.,
prostaglandin biosynthesis38). These model compounds have been
prepared directly from fatty acid precursors39 or from cyclopenta-1,3-dienes
followed by reduction of the remaining
C–C double bond.40
For most preparative purposes, however,
the photo-oxidation reactions have been designed in
such as way that either in situ or subsequent treatment of the
peroxides formed initially leads to synthetically valuable building
blocks for organic synthesis. Furthermore, the endoperoxides
are versatile starting materials for further transformations.
Nucleophilic ring opening is another possibility for
selective transformations that include reductions to diols, oxidations
to dicarbonyl products and rearrangements to hydroxy
carbonyl compounds, epoxy carbonyl compounds, bisepoxides,
ene diones, etc.
The
tris(2,2′-bipyridine)ruthenium(II) complex [Ru(bpy)32+] is a well known photosensitizer
for water oxidation and photo-reduction reactions.41–44
In addition, this complex can produce O2(1Δg) with
high quantum yield,8,45,46
comparable to those with Rose Bengal,
Methylene Blue, porphyrins and phthalocyanines. On the other hand, we have reported that partially quaternized
poly(1-vinylimidazole)-bound ruthenium(II) complexes (RuQPIms) are excellent photosensitizers for quenching of viologens,47
photosensitized charge separation48,49 and photoinduced
hydrogen generation.50–52
However, these polymer photosensitizers
have not
been applied to the photo-oxidation reaction.
In this paper, we report the first application of RuQPIms
as photosensitizers to the photo-oxidation of cyclopenta-1,3-diene (CP) with singlet oxygen in ethanol and the effects
of the alkyl side chain length. We selected two RuQPIms with a hexyl group
(C6RuQPIm) and a hexadecyl group (C16RuQPIm)
as the alkyl side chains because they are more stable for visible light irradiation than the non-quaternized ruthenium(II)
complex-containing poly(10-vinylimidazole). Tris(2,2′-bipyridine)ruthenium(II)
[Ru(bpy)32+], is used as a low molecular weight model
compound.
Experimental
Materials
All
reagents used for the syntheses of complexes were of
analytical-reagent grade and were used without further purification.
Ru(bpy)32+ was prepared according to the literature.53 Partially
quaternized poly(1-vinylimidazole)-bound ruthenium(II) complexes (RuQPIms) were prepared by quaternization of poly(1-vinylimidazole)-bound
ruthenium(II) complex (RuPIm)54 with hexyl bromide
or hexadecyl bromide in methanol.47
The structure is
shown in Fig. 1. Cyclopenta-1,3-diene (CP) obtained by distillation
of dicyclopentadiene just before use was used in all measurements
owing to its instability. |
| | Fig. 1 Structure
of partially quaternized poly(1-vinylimidazole)-bound ruthenium(II) complexes. | |
Measurements
UV–Vis absorption spectrum measurements were carried out using
a Perkin-Elmer Lambda 9 spectrophotometer. The photo-oxidation
experiments were performed at 25°C in 50 ml of
oxygen-saturated ethanol containing the photosensitizer and CP
using an automatic apparatus as described previously.17,19
The oxygen consumption was followed continuously
during stirring and irradiation with visible light from halogen
lamp (250 W). After bubbling of the ethanol solution containing
the photosensitizer for 10 min with oxygen, various concentrations
of the CP were added. The reactions were monitored
by measuring the oxygen consumption during the irradiation
period. The initial rate of the photo-oxidation reaction
was calculated from the initially linear slope of the graph
of oxygen consumption vs. irradiation time. The stability of these photosensitizers was followed by visible spectroscopy.
The absorbances of the photosensitizers in the reaction solutions,
which were 450 nm for Ru(bpy)32+ and 490 nm for RuQPIms,
before and after photo-oxidation were compared. Furthermore,
the stability was also evaluated by repeated experiments.
The repeated experiments were performed as follows. After bubbling of an ethanol solution of 5.0×10−5 M photosensitizer
for 10 min with oxygen, CP (2.5×10−2 M) was added and
then the oxygen consumption with visible light irradiation
was monitored during the irradiation period. The reaction solution
was bubbled with oxygen for 10 min again, and after the
addition of 2.5×10−2 M CP, the oxygen consumption was monitored.
This procedure was repeated. In all systems, the experimental
errors were within 0.5%.Results
When an oxygen saturated ethanol solution containing photosensitizer
and CP was irradiated with visible light, the oxygen
in the reaction cell was consumed (Fig.
2). Since the oxygen
was not consumed in the absence of the photosensitizer and
the quenching reaction of the photoexcited photosensitizers
with CP did not take place under an argon atmosphere, the
CP would be oxidized by singlet oxygen generated by energy
transfer from the photoexcited photosensitizer to triplet
molecular oxygen. The total amount of oxygen consumed was
ca. 1.11×10−3 mol for Ru(bpy)32+ and ca. 1.25×10−3 mol for these
polymers. Since this reaction cell contains
1.25×10−3
mol of CP, the CP reacts with an equivalent of singlet
oxygen generated by photoexcited photosensitizers for the polymer
systems. |
| | Fig. 2 Oxygen
consumption in oxygen-saturated ethanol solution containing 5.0×10−5 M photosensitizer and 2.5×10−2 M CP with visible light irradiation for Ru(bpy)32+, C6RuQPIm and C16RuQPIm systems. | |
Ru(bpy)32+ as photosensitizer
Fig. 3 shows the oxygen consumption in ethanol solution containing
various concentrations of Ru(bpy)32+ and 2.5×10−2
M CP
during visible light irradiation, together with the dependence
of the initial reaction rate on the concentration of Ru(bpy)32+.
For all concentrations of Ru(bpy)32+ in this experiment,
the total amount of oxygen consumed was 1.11×10−4
mol, although the reaction time when saturation of the
oxygen consumption was reached was different. The degree
of reaction, given by the molar ratio of initial CP and reacted
CP concentrations,55 was ca. 90%. The initial reaction rate evaluated
from the linear slopes of oxygen consumption with time
linearly increased with increase in the concentration of Ru(bpy)32+, and then remained constant above 4.0×10−5 M of Ru(bpy)32+. Therefore, subsequent experiments were carried out
with [Ru(bpy)32+]=5.0×10−5 M.![Oxygen
consumption in oxygen-saturated ethanol solution containing various concentrations of Ru(bpy)32+ and 2.5×10−2 M CP with visible light irradiation: [Ru(II
)]=1, 4.94×10−5; 2, 4.01×10−5; 3, 3.05×10−5; 4, 2.00×10−5; 5, 9.79×10−6; 6, 4.73×10−6 and 7, 2.12×10−6 M. Inset: dependence of initial reaction rate on concentration of Ru(bpy)32+.](/image/article/2000/CP/a906816c/a906816c-f3.gif) |
| | Fig. 3 Oxygen
consumption in oxygen-saturated ethanol solution containing various concentrations of Ru(bpy)32+ and 2.5×10−2 M CP with visible light irradiation: [Ru(II
)]=1, 4.94×10−5; 2, 4.01×10−5; 3, 3.05×10−5; 4, 2.00×10−5; 5, 9.79×10−6; 6, 4.73×10−6 and 7, 2.12×10−6 M. Inset: dependence of initial reaction rate on concentration of Ru(bpy)32+. | |
Fig. 4 shows the oxygen consumption in oxygen-saturated ethanol
solution containing 5.0×10−5 M Ru(bpy)32+ and various
concentrations of CP during visible light irradiation, together
with the dependence of the initial reaction rate on the concentration
of CP. The reaction rates, the total amount of oxygen
consumed and degree of reaction are summarized in Table
1. The photo-oxidation reaction was completed within 15 min
for all systems. The total amount of oxygen consumed and the
degree of reaction increased with increase in the initial concentration
of CP. The degree of reaction at the low CP concentrations
was very low; in particular, that at [CP]=7.5×10−3
and 5.0×10−3 M was ca. 62 and 54%, respectively.
The reaction rate increased linearly with increase in the concentration
of CP, and then remained constant above 2.0×10−2 M CP.
![Oxygen
consumption in oxygen-saturated ethanol solution containing 5.0×10−5 M Ru(bpy)32+ and various concentrations of CP with visible light irradiation: [CP]=1, 2.5×10−2; 2, 2.0×10−2; 3, 1.5×10−2; 4, 1.0×10−2; 5, 7.5×10−3 and 6, 5.0×10−3 M. Inset: dependence of initial reaction rate on concentration of CP.](/image/article/2000/CP/a906816c/a906816c-f4.gif) |
| | Fig. 4 Oxygen
consumption in oxygen-saturated ethanol solution containing 5.0×10−5 M Ru(bpy)32+ and various concentrations of CP with visible light irradiation: [CP]=1, 2.5×10−2; 2, 2.0×10−2; 3, 1.5×10−2; 4, 1.0×10−2; 5, 7.5×10−3 and 6, 5.0×10−3 M. Inset: dependence of initial reaction rate on concentration of CP. | |
Table 1
Initial reaction rate (R) and total amount of oxygen consumed (OC) for photo-oxidation of CP using the photosensitizersa
| Ru(bpy)32+ | C6RuQPIm | C16RuQPIm |
|---|
| | | |
|---|
| [CP]/10−2M | R/10−6 mol s−1 | OC/10−4 mol | R/10−6 mol s−1 | OC/10−4 mol | R/10−6 mol s−1 | OC/10−4 mol |
|---|
| |
|---|
| Concentration of photosensitizers is ca. 5.0×10−5 M. Degree of reaction (%). |
|---|
| 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.50 | 0.83 | 1.34(53.6b) | 0.89 | 2.21(88.4b) | 1.59 | 2.5(100b) |
| 0.75 | 1.42 | 2.32(61.9b) | 1.46 | 3.53(94.1a) | 1.85 | 3.75(100b) |
| 1.00 | 2.05 | 4.21(84.2b) | 1.67 | 4.53(90.6b) | 1.87 | 5.02(100b) |
| 1.50 | 2.52 | 6.12(81.6b) | 1.69 | 7.49(99.9b) | 1.93 | 7.51(100b) |
| 2.00 | 2.66 | 8.14(81.4b) | 1.67 | 10.1(100b) | 1.97 | 9.99(100b) |
| 2.50 | 2.77 | 11.1(88.8b) | 1.44 | 12.44(100b) | 1.96 | 12.49 (100b) |
Polymer photosensitizer systems
Fig. 5 shows the oxygen consumption in oxygen-saturated ethanol
solution containing 2.5×10−2 M CP and various
concentrations
of the polymer photosensitizer, together with the dependence of the initial reaction rate on the concentration
of the ruthenium(II) complex. For all concentrations of ruthenium(II)
complex in these experiments, the total amount of oxygen consumed was ca.
1.25×10−4 mol and the degree of reaction was almost 100%. The
initial reaction rate increased linearly with increase
in the ruthenium(II) complex concentration, and then remained constant
above 4.0×10−5 M Ru(II) complex for all systems. Therefore,
subsequent experiments were carried out with [Ru(II)]=5.0×10−5 M.![Oxygen
consumption in oxygen-saturated ethanol solution containing various concentrations of (A) C6RuQPIm or (B) C16RuQPIm and 2.5×10−2 M CP with visible light irradiation: [Ru(II
)]=1, 5.03×10−5; 2, 4.04×10−5; 3, 3.05×10−5; 4, 2.03×10−5; 5, 9.88×10−6; 6, 4.94×10−6 and 7, 2.08×10−6 M. Inset: dependence of initial reaction rate on concentration of the ruthenium(II
) complex.](/image/article/2000/CP/a906816c/a906816c-f5.gif) |
| | Fig. 5 Oxygen
consumption in oxygen-saturated ethanol solution containing various concentrations of (A) C6RuQPIm or (B) C16RuQPIm and 2.5×10−2 M CP with visible light irradiation: [Ru(II
)]=1, 5.03×10−5; 2, 4.04×10−5; 3, 3.05×10−5; 4, 2.03×10−5; 5, 9.88×10−6; 6, 4.94×10−6 and 7, 2.08×10−6 M. Inset: dependence of initial reaction rate on concentration of the ruthenium(II
) complex. | |
Fig. 6 shows the oxygen consumption in oxygen-saturated ethanol
solution containing 5.0×10−5 M Ru(II) complex and various concentrations
of CP during visible light irradiation, together with the
dependence of the initial reaction rate on the concentration of
CP. The photo-oxidation reaction was completed within 20
min and the degree of reaction was very high for these systems.
In particular, the C16RuQPIm systems demonstrated 100%
reaction even when the CP concentration was low. The reaction
rate depends significantly on the length of the alkyl side
chain. For the polymer systems, the reaction rate increased up
to [CP]=1.0×10−2
M and then remained almost constant.
![Oxygen
consumption in oxygen-saturated ethanol solution containing 5.0×10−5 M polymer photosensitizer and various concentrations of CP with visible light irradiation: [CP]=1, 2.5×10−2; 2, 2.0×10−2; 3, 1.5×10−2; 4, 1.0×10−2; 5, 7.5×10−3; and 6, 5.0×10−3 M. Inset: dependence of initial reaction rate on concentration of CP.](/image/article/2000/CP/a906816c/a906816c-f6.gif) |
| | Fig. 6 Oxygen
consumption in oxygen-saturated ethanol solution containing 5.0×10−5 M polymer photosensitizer and various concentrations of CP with visible light irradiation: [CP]=1, 2.5×10−2; 2, 2.0×10−2; 3, 1.5×10−2; 4, 1.0×10−2; 5, 7.5×10−3; and 6, 5.0×10−3 M. Inset: dependence of initial reaction rate on concentration of CP. | |
Stability of photosensitizers
The stability of the photosensitizers was evaluated by repeated
experiments on the photo-oxidation reaction and the absorption spectra before and after irradiation. Fig. 7 shows the repeated
experiments on the photo-oxidation reaction of CP using
(A) Ru(bpy)32+, (B) C6RuQPIm and (C) C16RuQPImas photosensitizers
at [Ru(II)]=5.0×10−5 M and [CP]=2.5×10−2
M. During the
repeated experiments using Ru(bpy)32+ photosensitizer,
the reaction was completed in all cycles within 15 min and the
total amount of oxygen consumed was ca. 1.11×10−4 mol;
in addition, the initial reaction rate did not change. Furthermore,
the absorption
spectrum measurements showed
that the absorbance and maximum wavelength did not change
during the repeated experiments; therefore, the
Ru(bpy)32+ photosensitizer was very stable.![Repeated
experiments of photo-oxidation reaction of CP using (A) Ru(bpy)32+, (B) C6RuQPIm and (C) C16RuQPIm as photosensitizers at [Ru(II
)]=5.0×10−5
M and [CP]=2.5×10−2 M, with the number of runs in ethanol on the abscissa. The irradiation time of one cycle is 15 min for the Ru(bpy)32+ system and 20 min in 1st to 9th cycles and 10th and 11th cycles for polymer systems.](/image/article/2000/CP/a906816c/a906816c-f7.gif) |
| | Fig. 7 Repeated
experiments of photo-oxidation reaction of CP using (A) Ru(bpy)32+, (B) C6RuQPIm and (C) C16RuQPIm as photosensitizers at [Ru(II
)]=5.0×10−5
M and [CP]=2.5×10−2 M, with the number of runs in ethanol on the abscissa. The irradiation time of one cycle is 15 min for the Ru(bpy)32+ system and 20 min in 1st to 9th cycles and 10th and 11th cycles for polymer systems. | |
On the other hand, for the polymer photosensitizer systems,
the initial reaction rate and the amount of oxygen consumption
after irradiation for 20 min decreased with repeated light
on-off cycles. Up to the fourth cycle, the amount of oxygen consumed
was the same, indicating that the reaction was completed
in 20 min. From the fifth to tenth cycles, however, the amount
of oxygen consumed decreased, and the reaction was incomplete.
When the irradiation was carried out for 30 min, the
amount of oxygen consumed was the same in the first and second
cycles. In the differential absorption spectra after irradiation
for various cycles,56 the absorbance around 510 nm increased
with repeated light on-off cycles.
Discussion
Ru(bpy)32+
and polymer photosensitizer systems
Fig.
8 shows the dependence of the initial reaction rate on the
concentration of photosensitizer (A) and CP (B) for
Ru(bpy)32+ and polymer photosensitizer systems. The initial reaction
rate for the Ru(bpy)32+ system is similar to that for the
polymer systems at a low photosensitizer concentration, whereas
it is larger at a high photosensitizer concentration. The Ru(bpy)32+ photosensitizers do not interact with each other
under the present experimental conditions. In contrast, it
is known that these photosensitizers undergo an inter-polymer
interaction between the alkyl side chains at high concentration,57–60
hence, these polymer photosensitizers would form
a heterogeneous reaction field where the photo-oxidation
reaction takes place. In the low photosensitizer concentration
range, the photo-oxidation reaction for both Ru(bpy)32+
and polymer systems takes place in the homogeneous reaction
field, consequently leading to similar reaction rates. In
the high photosensitizer concentration range, the photo-oxidation reaction occurs in the homogeneous field for the Ru(bpy)32+ system, but the polymer photosensitizers provide a
heterogeneous field for the reaction, giving a low initial reaction
rate for the polymer systems. Furthermore, the inactivation of part of the ruthenium(II) complexes is a very important factor. With
increasing concentration of the ruthenium(II) complex,
the luminescence intensity increases linearly, and reaches a saturation
value at high concentration, probably owing to self-quenching between
the ruthenium(II) complexes.![(A)
Dependence of initial reaction rate on concentration of ruthenium(II
) complex at [CP]=2.5×10−2 M. (B) Dependence of initial reaction rate on concentration of CP at [Ru(II
)]=5.0×10−5
M: (●) Ru(bpy)32+; (■) C6RuQPIm; (▲) C16RuQPIm.](/image/article/2000/CP/a906816c/a906816c-f8.gif) |
| | Fig. 8 (A)
Dependence of initial reaction rate on concentration of ruthenium(II
) complex at [CP]=2.5×10−2 M. (B) Dependence of initial reaction rate on concentration of CP at [Ru(II
)]=5.0×10−5
M: (●) Ru(bpy)32+; (■) C6RuQPIm; (▲) C16RuQPIm. | |
As shown in Fig. 8(B), the CP concentration dependence of
the initial reaction rate for the Ru(bpy)32+ system is similar to the photosensitizer concentration dependence, but not for the polymer systems. This is the reason why these systems differ in the
reaction field. The initial reaction rate for the Ru(bpy)32+
system
is higher than that for the polymer systems at high CP concentration,
but is lower than that for the polymer systems at
low CP concentration. In particular, the initial reaction rate for
C16RuQPIm system at [CP]=5.0×10−3 M is double that for
the Ru(bpy)32+ system. This result is attributed to a concentration
effect induced by incorporation of the CP species
into the heterogeneous field formed by these polymer photosensitizers,
as illustrated in Scheme 1.
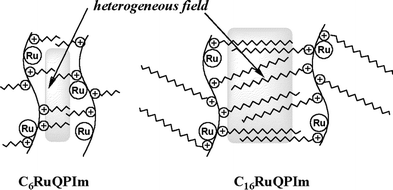 |
| | Scheme 1 Tentative representation of heterogeneous reaction field formed by polymer photosensitizers. | |
The heterogeneous reaction field, in which the alkyl side
chains are closely packed, would have solvophobic properties,
and is able to concentrate CP at low CP concentration. Consequently,
the C16RuQPIm system demonstrates a high reaction
rate at low CP concentration. Furthermore, the initial
reaction rate is almost constant regardless of the CP concentration
even when the latter increases, because the amount of
CP which the heterogeneous reaction field can incorporate is
limited. In addition, the fact that the degree of reaction for the
polymer photosensitizer systems is almost 100% can be also explained
by the same reasoning.
Effect
of alkyl side chain length
Although
the polymer photosensitizers form a heterogeneous
reaction field, the solvophobic properties and the amount of
CP incorporated into the field depend significantly on the
length of the alkyl side chain of the polymers. The C6RuQPIm
photosensitizer with short alkyl side chains forms a heterogeneous
reaction field which has a low solvophobicity and a
small size owing to electrostatic repulsion between the
polymer chains and the small van der Waals interaction
between the alkyl side chains. In contrast, the heterogeneous
reaction field formed by the C16RuQPIm photosensitizer has
a high solvophobicity and a large size. The steric hindrance of
the long alkyl side chains decreases the electrostatic repulsion
and increases the size of the field. In addition, the long alkyl
side chains, which induce strong van der Waals interactions,
bring about high solvophobicity, therefore leading to an
increase in the amount of CP incorporated into the field.Stability
of photosensitizers
As
mentioned above, the Ru(bpy)32+
photosensitizer demonstrates
excellent stability. During repeated experiments on the photo-oxidation
reaction, the initial reaction rate and saturated
oxygen consumption hardly change. Furthermore, no change
in the absorption spectra with visible light irradiation is
observed.Fig.
9 shows the change in the absorption spectrum of
C16RuQPIm during the repeated experiments, together with
differential absorption spectral change.61 Clearly, a red shift of
the absorption maxima is observed, and the absorbance at 510
nm in the differential spectra increases, which indicates that
these polymer photosensitizers change to the different complexes. It is well known that the bis(2,2′bipyridine)chloro-N-methylimidazolylruthenium(II) complex, [Ru(bpy)2(MeIm)(Cl)]2+, has
an absorption maximum at 512 nm and luminescence maxima
at 690 nm in methanol.54
Con
sidering the fact, it appears
that the polymer photosensitizers change to monochloro complexes
[CnRu(Cl)QPIm (n=6 or 16)] in which one imidazolyl
residue is substituted by a chloride ion that is the counter
ion of the ruthenium(II) complex.62 Therefore, the decrease in reaction activity
is caused by the formation of CnRu(Cl)QPIm. Although the
CnRu(Cl)QPIm photosensitizers have a low reaction activity
comparable to that of the original complexes (CnRuQPIms),
they demonstrate the same maximum oxygen consumption
as that of CnRuQPIms when the irradiation time is varied
up to 30 min. Moreover, it is confirmed by the repeated
experiments that the CnRu(Cl)QPIm photosensitizers also have
excellent stability for the photo-oxidation reaction.
 |
| | Fig. 9 Change
in absorption spectrum of C16RuQPIm during the repeated experiments. Inset: differential absorption spectral change. | |
Comparison with other photosensitizers
In order to make a comparison with other photosensitizers,
the photo-oxidation reaction was carried out using Rose Bengal (RB) and zinc(II) phthalocyaninetetrasulfonic acid (ZnPTS) as the
photosensitizers. These photosensitizers demonstrate excellent
activity for the photo-oxidation reaction. The initial
reaction rates are much larger than those of ruthenium(II) complex photosensitizers under the same conditions in the ruthenium(II)
complex photosensitizer systems,63 and the reaction is completed within 10
min even when the RB and ZnPTS concentrations are 2.5×10−6
M. As shown in Fig.
10, however, the stability of these
photosensitizers is very low: more than 50% of RB and ZnPTS has
decomposed after the reaction. This is the reason why
RB and ZnPTS are
attacked by singlet oxygen.64
Furthermore,
the degree of reaction for RB and ZnPTS is ca. 85%, which is
similar to that of Ru(bpy)32+. These results indicate that the
Ru(bpy)32+ and polymer-bound ruthenium(II) complexes have
excellent stability comparable to that of RB and ZnPTS, although the reaction
activity is low.![Absorption
spectra of RB (dotted lines) and ZnPTS (solid lines) before and after the reaction at [photosensitizer]=2.5×10−6 M and [CP]=2.5×10−2 M.](/image/article/2000/CP/a906816c/a906816c-f10.gif) |
| | Fig. 10 Absorption
spectra of RB (dotted lines) and ZnPTS (solid lines) before and after the reaction at [photosensitizer]=2.5×10−6 M and [CP]=2.5×10−2 M. | |
Conclusion
We have carried out the photo-oxidation of cyclopenta-1,3-diene
(CP) with singlet oxygen using Ru(bpy)32+ and polymer-bound ruthenium(II) complexes (C6RuQPIm and C16RuQPIm) as the
photosensitizers in oxygen-saturated ethanol. These ruthenium(II)
complex photosensitizers have excellent stability comparable
to those of other photosensitizers such as Rose Bengal
and ZnPTS, and can be repeatedly used for photo-oxidation.
The polymer photosensitizer systems can oxidize
CP more effectively than the Ru(bpy)32+ system. In particular,
the C16RuQPIm system oxidizes all of the CP added even
when the CP concentration is low, i.e., these polymer photosensitizers
are effective at low concentrations of CP. This is attributed
to the fact that the polymer photosensitizers form a
heterogeneous reaction field in which CP species can be concentrated.
Furthermore, these polymer photosensitizers can be
separated and recovered from the reaction solution using a simple
treatment, by precipitation through addition of acetone.
In addition, the CnRu(Cl)QPIms return to the original
complexes, CnRuQPIms, on refluxing in methanol.References
- I.
M. Byteva and G. P. Gurinovich, J. Lumin., 1979, 21, 17 CrossRef CAS.
- J. R. Hurst, J. D. MacDonald and G. D. Schuster, J. Am. Chem. Soc., 1982, 104, 2065 CrossRef CAS.
- J. G. Parker and W. G. Stanbro, J. Am. Chem. Soc., 1982, 104, 2067 CrossRef CAS.
- P. R. Ogilby and C. S. Foote, J.
Am. Chem. Soc., 1982, 104, 2069 CrossRef CAS.
- M. A. J. Rodgers and P. T. Snowden, J. Am. Chem. Soc., 1982, 104, 5541 CrossRef CAS.
- G. Rossbroich, N. A. Garcia and S. E. Braslavsky, J. Photochem., 1985, 31, 37 Search PubMed.
- R. W. Redlmond, Photochem.
Photobiol., 1991, 54, 547, and references cited therein Search PubMed.
- W. Spiller, H. Kliesch, D. Wöhrle, S. Hackbarth, B. Röder and G. Schnurpfeil, J. Porphyrins Phthalocyanines, 1998, 2, 145 CrossRef CAS.
- J.
D. Spikes, J. Photochem. Photobiol., B, 1990, 6, 259 Search PubMed.
- B. Röder and D. Nähter, Proc. SPIE, 1991, 1525, 377 Search PubMed.
- M. O.
K. Obochi, A. J. Canaan, A. K. Jaen, A. M. Richter and J. G. Levy, Photochem. Photobiol., 1992, 62, 169 Search PubMed.
- J. M. Wessels, W. Strauss, H. K. Seidlitz, A. Rück and H. Schneckenberger, J. Photochem. Photobiol., B, 1992, 12, 275 Search PubMed.
- L. Roitman, B. Ehrenberg and N. Kobayashi, J. Photochem. Photobiol., A, 1994, 77, 23 Search PubMed.
- P. F. Aramendia, R. W. Redmond, S. Nonell, W. Schuster, S. E. Braslavsky, K. Schaffner and E. Vogel, Photochem. Photobiol., 1986, 44, 555 Search PubMed.
- I. Rosenthal, Photochem. Photobiol., 1991, 53, 859 Search PubMed.
- T. J. Dougherty, Adv. Photochem., 1992, 17, 275 Search PubMed.
- W. Spiller, D. Wöhrle, G. Schulz-Ekloff, W. T. Ford, G. Schneider and J. Stark, J. Photochem. Photobiol., A, 1996, 95, 161 Search PubMed.
- D. Wöhrle, W. Spiller, G. Schneider, G. Schulz-Ekloff and J. Stark, J. Inf. Rec. Mater., 1994, 21, 1 Search PubMed.
- G. Schneider, D. Wöhrle, W. Spiller, J. Stark and G. Schulz-Ekloff, Photochem. Photobiol., 1994, 60, 333 Search PubMed.
- D. Wöhrle, G. Schneider, J. Stark and G. Schulz-Ekloff, J. Mol. Catal., 1992, 75, L39 CrossRef.
- H. E. Gsponer, C.
M. Previtali and N. A. Garcia, Toxicol. Environ. Chem., 1987, 16, 23 Search PubMed.
- M. C. Palumbo and N. A. Garcia, Toxicol. Environ. Chem., 1988, 17, 103 Search PubMed.
- R. Gerdes, D. Wöhrle, W. Spiller, G. Schneider, G. Schnurpfeil and G. Schulz-Ekloff, J. Photochem.
Photobiol., A, 1997, 111, 65 Search PubMed.
- R. C. Straight
and J. D. Spikes,
in Singlet Oxygen, ed. A. A. Frimer, CRC
Press, Boca Raton, FL, 1985, vol. IV, p. 91. Search PubMed.
- J. R. Wagner, H. Ali, R. Langlois, N. Brasseur and J. E. van Lier, Photochem. Photobiol., 1987, 45, 587 Search PubMed.
- J.-L. Ravanat, M. Berger, F. Benard, R. Langlois, R. Ouellet, J.
E. van Lier and J. Cadet, Photochem.
Photobiol., 1992, 55, 809 Search PubMed.
- S. Rywkin, L. Lenny, J. Goldstein, N. E. Geacintov, H. Marglois-Nunno and B. Horowitz, Photochem. Photobiol., 1992, 56, 463 Search PubMed.
- X.-F. Zhang and H.-J. Xu, J. Photochem. Photobiol.,
B, 1994, 24, 109 Search PubMed.
- J. D. Spikes, J. E. van Lier and J. C. Bommer, J. Photochem. Photobiol.,
A, 1995, 91, 193 Search PubMed.
- A. A. Gorman, G. Lovering and M. A. J. Rodgers, J. Am. Chem.
Soc., 1979, 101, 3050 CrossRef CAS.
- B. M. Monroe, J. Am. Chem. Soc., 1981, 103, 7253 CrossRef CAS.
- G. O. Schenck and D. E. Dunlap, Angew. Chem., 1956, 68, 248 CrossRef CAS.
- K. H. Schulte-Elte, B. Willhalm and G. Ohlof, Angew. Chem., 1969, 81, 1045.
- G. O. Schenck, Angew. Chem., 1952, 64, 12 CAS.
- A. C. Cope, T. A. Liss and G. A. Wood, J. Am. Chem. Soc., 1957, 79, 6287 CrossRef CAS.
- A. Horinaka, R. Nakashima, M. Yoshikawa and T. Matsuura, Bull. Chem. Soc.
Jpn., 1975, 48, 2095 CAS.
- P. Esser, B. Pohlmann and H.-D. Scharf, Angew. Chem., 1994, 106, 2093 CAS.
- M. Hamberg and B. Samuelson, J. Biol. Chem., 1967, 242, 5336 CAS.
- E. D. Mihelich, J.
Am. Chem. Soc., 1980, 102, 7141 CrossRef CAS.
- W. Adam and H. J. Eggelte, J. Org. Chem., 1977, 42, 3987 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159, and references
cited
therein CrossRef.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von
Zelewsky, Coord. Chem. Rev., 1988, 84, 85,
and references cited therein CrossRef.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141,
and references
cited therein CAS.
- W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1,
and references cited therein CrossRef CAS.
- K. Bhattacharyya and P. K. Pas, Chem. Phys. Lett., 1985, 116, 326 CrossRef CAS.
- R. A. Kenley, N. A. Kirshen and T. Mill, Macromolecules, 1980, 13, 808 CAS.
- M. Suzuki, Y. Mori, N. Yokoyama, M. Kimura, K. Hanabusa and H. Shirai, Macromol. Chem. Phys., 1997, 198, 959 CrossRef CAS.
- M. Suzuki, Y. Mori, M. Kimura, K. Hanabusa and H. Shirai, J. Chem. Soc., Faraday Trans., 1996, 92, 3599 RSC.
- M. Suzuki, M. Kimura, K. Hanabusa and H. Shirai, J. Chem. Soc., Faraday
Trans., 1997, 93, 4173 Search PubMed.
- M. Suzuki, S. Kobayashi, S. Uchida, M. Kimura, K. Hanabusa and H. Shirai, Chem. Commun., 1997, 227 RSC.
- M. Suzuki, M. Kimura, K. Hanabusa and H. Shirai, Macromol.
Chem. Phys., 1998, 199, 945 CrossRef CAS.
- M. Suzuki, S. Kobayashi, S. Uchida, M. Kimura, K. Hanabusa and H. Shirai, Polymer, 1998, 39, 1539 CrossRef CAS.
- P. A. Palmer and T. S. Piper, Inorg. Chem., 1966, 5, 864 CrossRef.
- S. M. Geraty and J. G. Vos, J. Chem. Soc., Dalton Trans., 1987, 3073 RSC.
-
In this case,
the reaction cell (50 ml) contains 12.5×10−4 mol of CP..
-
Differential
absorption spectra after irradiation were defined as the spectra
obtained by
the subtraction of the spectrum before irradiation from that after
irradiation..
- M. Suzuki, M. Yokoyama, M. Kimura, K. Hanabusa and H. Shirai, Eur. Polym. J., 1999, 35, 1057 CrossRef CAS.
- M. Suzuki, M. Sano, M. Kimura, K. Hanabusa and H. Shirai, Eur. Polym. J., 1999, 35, 1079 CrossRef CAS.
- M. Suzuki, M. Yokoyama, M. Kimura, K. Hanabusa and H. Shirai, React. Funct. Polym., 1999, 40, 241 CrossRef CAS.
-
In the present system, the
inter-polymer
interaction was confirmed by viscosity measurements.
In the low concentration range of the ruthenium(II
) complex, the viscosity increased with increasing concentration
of the ruthenium(II
) complex, whereas it
decreased in the high concentration range..
-
The C6RuQPIm system
showed the same result as the C16RuQPIm
system..
-
The luminescence spectra of the solution after
the reaction was completed showed a luminescence
maximum at 692 nm, corresponding to the monochloro complex
species, and the intensity
increased during the repeated experiments..
-
Under the conditions
of [RB] or [ZnPTS]=2.5×10−6 M and [CP]=2.5×10−2
M, the reaction rates are 1.66×10−6 mol s−1 for RB and 1.79×10−6
mol s−1 for ZnPTS..
- G. Schnurpfeil, A. K. Sobbi, W. Spiller, H. Kliesch and D. Wöhrle, J. Porphyrins Phthalocyanines, 1997, 1, 159 CrossRef CAS.
Footnote |
| † Research
Fellow of the Japan Society for the Promotion of Science
(JSPS Research Fellow). |
|
| This journal is © the Owner Societies 2000 |
Click here to see how this site uses Cookies. View our privacy policy here.